Afrocentric Melanist Theorists
2650 words
Extremists exist in every ideology (there are too many to name but take the books March of the Titans (Kemp, 1999) to Testosterone Rex (Fine, 2017) as examples), but some are (in my opinion) more extreme (and funnier and more delusional) than others (even if they’re almost neck and neck). Afrocentrists veer toward the extreme side (as Nordicists veer to the extreme side on the opposite end). But certain beliefs these ideologies have may “sound right” to the uneducated ear, especially when they begin to weave fantastical stories with physiological terminology in order to woo the listener. You see things like “Melanin affects the idea of white supremacy“, but what does this really mean (you will see what it means near the end of this article)? The one saying it may believe it themselves, though it all doesn’t make sense. The extremist views that are more interesting are the Afrocentric ones, though, especially the melanin theory that gets thrown around in Afrocentric circles.
Melanin production
Melanin is produced by melanocytes. Melanin is synthesized from L-tyrosine, with the help of tyrosinase, which is one of the main enzymes for melanin production (Solano, 2014; D’Mello et al, 2016). (See Cone, 2006 for a review of the melanocortin system.) Melanin absorbs energy from UV rays which then dissipate in the body as heat (de Monteallano, 1993). There are three types of melanin: eumelanin (there are two types of eumelanin: brown eumelanin and black eumelanin), pheomelanin (these two are present in the human epidermis; Thody et al, 1991; Solano, 2014) and neuromelanin. Pheomelanin and eumelanin are found in the hair and skin.
Races that live closer to the equator have higher concentrations of melanin in their skin (not neuromelanin, which will be discussed later) which then causes dark skin pigmentation. But everyone on earth has around the same number of melanocytes; skin pigmentation differences come down to differences in UV exposure (for which melanin is useful; and produced due to UV radiation Brenner and Hearing, 2008), disease (albinism and vitiligo), size of melanocytes (see below) and genetic make up.
Europeans and Chinese have about half as much melanin as African and Indian skin types, whereas Africans had the largest melanosomes, followed by the Indians, Mexicans, Chinese, and Europeans, therefore variation in melanosome size may also account for skin variation between races. It’s also interesting to note that people, no matter the skin color, who are born in high UV areas—regardless of ethnicity—have twice as much epidermal melanin compared to people born in low UV areas (Alaluf et al, 2002).
Melanin and pseudoscience
Rushton and Templer (2012) wrongly hypothesized that the melanocortin system modulated sexuality and aggression and humans as they do in animals. The claims made here are may “sound good” to one who isn’t well-versed in the physiology of aggression and sexuality, but to those people, Rushton and Templer’s hypothesis “sounds good enough” and so they believe it without question. On the opposite side, you have black “academics” who believe that melanin gives blacks some type of “greatness” and is the reason for their “natural moves.’ In the book Darwin’s Athletes: How Sport Has Damaged Black America and Preserved the Myth of Race, Hoberman (1997: 89) shortly discusses Afrocentric melanin theorists:
Finally, there are the melanin theorists. a motley collection of pseudo-scientific cranks and better-known members of the black academic demimonde who attended the Fourth Annual World Melanin Conference in Dallas in April 1989—Leonard Jeffries, John Henrik Clarke, Ivan Van Sertima, and others. For these racial biologists, the pigment that makes skin dark is “the Chemical Key to Black Greatness” and accounts for an entire range of superior black aptitudes: “The reason why Black athletes do so well and have these ‘natural moves’ is these melanic tracks in the brainstem tie into the cerrebellum . . . a part of us that controls motor movement (Dr. Richard King). The real signifigance of the melanin theory is that it is the reductio ad absurdum of black racial seperatism, putting its adherents in a de facto alliance with white racists, who have their own reasons to establish separate racial physiologies. Afrocentric science curricula that promote melanin theory have been introduced in a nimber of urban school districts in the United States, thereby doing educational damage to those children who can least afford it.
Note how there are similarities to Rushton and Templer’s (2012) hypothesis on the melanocortin system in darker-pigmented races (mainly blacks since that’s the race they theorized on). But what I find the funniest about melanin theory, as that some Afrocentrists use higher levels of melanin as “physiologic” proof that blacks are “superior athletes” (this can be explained without appealing to melanin). Though do note how the Afrocentric view of melanin and Rushton and Templer’s (2012) view of the melanocortin system and melanin are stark opposites of each other—and they’re both horribly wrong.
Now let’s look at some quotes from some Afrocentric websites.
These quotes are from Suzar’s (1999) book Blacked Out Through Whitewash: Exposing the Quantum Deception/Rediscovering and Recovering Suppressed Melanated (the author cites another book, Melanin: The Chemical Key to Black Greatness by Carol Barnes (1988):
“…your mental processes (brain power) are controlled by the same chemical that gives Black humans their superior physical (athletics, rhythmic dancing) abilities. This chemical…is Melanin!”
Then writing:
The abundance of melanin in Black humans produces a superior organism both mentally and physically. Black infants sit, stand, crawl and walk sooner than whites, and demonstrate more advanced cognitive skills than their white counterparts because of their abundance of melanin. Melanin is the neuro-chemical basis foe what is called “SOUL” in Black people. Melanin refines the nervous system in such a way that messages from the brain reach other areas of the body more rapidly in Black people than in the other. In the same way Blacks excel in athletics, Blacks can excel in all other areas as well (like they did in the past!) once the road blocks are removed.

Notice how this uses Rushton-like data similar to his ‘life history/r/K’ theory of human racial differences. People can have any kind of data they want, but when they start discussing the data then they are leaving the realm of science and are entering the realm of philosophy. They then interpret the data wrong, as evidence for ‘superiority’ in certain traits, and those who are less informed will buy it without question. Do note the similarities to Rushton and Templer’s (2012) hypothesis on the causes for sexual behavior and aggression differences in human races: melanin and the melanocortin system is partly a cause for these racial disparities. You only need a ‘good story’ (a just-so story) that seems like it is a plausible explanation in order to lure someone who’s unsuspecting to pseudo-science. (I would liken melanin to ‘g’ here. Both melanin and ‘g’ are given ‘powers’ that do not exist; but in the case of ‘g’, it doesn’t exist so at least Afrocentrists are discussing an actual hormone, though they are horribly misrepresenting what the actual data on melanin says.)
The most in-depth take-down of Afrocentric melanist theories is from de Mantellano (1993). Afrocentric theory states that black people—and Egyptians because they were black too (they weren’t)—since they have higher levels of melanin in their skin, then this gives them physical and mental superiority over those with less melanin in their skin. They misinterpret (willingly or not) many papers in order to push their pseudo-scientific theories to the ignorant masses (which already is occurring in inner-city schools, widening the already wide science gaps; see de Manteallno, 1992). What Afrocentrists do not understand is that all humans have similar anounts of neuromelanin (which they wrongly conflate with skin melanin), while neuromelanin levels in the brain are also independent of melanin levels in the skin. So the fantastic claims of melanin causing physical and mental ‘superiority’ (whatever that is) for darker-skinned individuals is unfounded.
Further claims from Afrocentrists are that since blacks have more skin melanin then this also means they have more melatonin and beta-melanocyte-stimulating hormone. Melatonin in humans also has no physiological relationship to skin color (de Mantellano, 1993). Lastly, Afrocentric melanists also state, as I have covered before, that Europeans are African albino mutants.
In fact, the claim that whites are just African albino mutants is ridiculous. Whites can produce eumelanin, while albinos can’t. Albinos are also homozygous recessive—since albinism is a Mendelian disorder, one must be homozygous recessive for “the albinism gene” (de Mantellano, 1993: 42). They can mate forever and they will never create offspring with the ability to synthesize melanin. Therefore it is impossible for whites to have been African albinos.
De Mantellano (1993) concludes that the theory is not to be taken seriously (of course) but states that “The idea that there are distinct races and that one is superior to the others is as racist and erroneous when it refers to high melanin levels as it was when it described low melanin levels (the Aryan “master race”)” (pg 54). Of course, no ‘master race’ exists, and the concept of ‘superiority’ has no basis in evolutionary biology, but race exists and is a biological reality. Though that doesn’t mean that any of the Afrocentric claims covered here have any basis—that’s because they conflate neuromelanin and melanin in the skin, even if they didn’t conflate the two they still would not be correct.
The fatal flaw in this type of Afrocentric “reasoning” is that neuromelanin differs in structure, location, and biosynthesis from skin melanin. Afrocentrists assert that neuromelanin and skin melanin are correlated. Though what falsifies this assertion is that albinos have the same amount of neuromelanin in their brains as non-albinos. So all of the purported ‘mental and physical superiority’ that was ’caused by melanin’ makes no sense, because neuromelanin and skin melanin were conflated. Neuromelanin does not even have the physiologic effects that most Afrocentrists believe.
Most Afrocentric melanists also cite individuals who cite…. rat studies and then extrapolate those results to humans. This is dumb. Yes I know the tired old “Humans are animals too!” but just because we’re animals too doesn’t mean that hormones work the same way in all species; it’s just some sort of bland appeal.
Perhaps one of the most amusing parts of de Mantellano (1993) is where he quotes a few prominent Afrocentrists who ‘argue’ that white men are afraid of black men because “Africans have very dominant genes”:
The conspiracy to destroy black youth. . . . It has to do with the fact that in terms of genetics and genes that because Africans have dominant genes that it is very possible for Africans to annihilate the European population. And the best way to prevent the annihilation is to get to the root of the perpetrator who could do that.
And that, of course, would be African men. Because it is men, specifically African men, that start the reproductive process off. For example, in looking at the four possibilities of sexual relationships. Of looking at those four there is only one possibility to produce a European child. If you have an African man with an African woman you will produce a child of color. If you have an African man with a European woman you will also produce a child of color. If you have a European man with an African woman that will also produce a child of color. European men can only produce a child that looks like them when they connect with a European woman. As the result of that, then, European men are very much afraid of African men and the conspiracy is directly centered at them. . . . And that’s that conspiracy is synonymous with the word genocide, and genocide not only is gradual, it is collective (Kunjufu, 1989).[…]
The reason that the Black male . . . is and always has been central to the issue of white supremacy is clarified by the definition of racism as white genetic survival. In the collective white psyche, Black males represent the greatest threat to white genetic survival because only males (of any color) can impose sexual intercourse, and Black males have the greatest genetic potential (of all non-white males) to cause white genetic annihilation. Thus, Black males must be attacked and destroyed in a power system designed to assure white genetic survival. . . . The prevention of white genetic annihilation is pursued through all means, including chemical and biological warfare. Today, the white genetic survival imperative, instead of using chemicals in gas chambers, is using chemicals in the streets-crack, cocaine, ecstasy, PCP, heroin and methadon [sic] (all “designer chemicals”). [Welsing, 1991a: 4]
Other more outlandish ideas are quoted by de Mantellano (1993) too, and all of the claims made about the physiology of melanin, neuromelanin bringing supernatural, physical and mental powers are horribly flawed. These people have no understanding of the physiology of the hormone, nor what they’re really speaking about. These attempted physiological theories to attempt to show racial ‘superiority’ make absolutely no sense if one has a basic understanding of the physiological system.
Claims made by Afrocentrists regarding melanin and neuromelanin range from blacks having more melanin in their muscle cells which is the cause for black athleticism; darker-eyed people having quicker reaction times which was thought to be caused by melanin; melanin centers in the brain being important for controlling and coordination of the body and brain power; to being critical for control of memory, motivation, mental maturation etc; causing altered states of consciousness which then causes black people who attend Church to speak in toungues; helps in the processing of memory; melanin and the pineal gland is at highest functionality in humans; and they conflate skin melanin with neuromelanin, when they are two different hormones (references for these claims can be found in de Montenallo, 1993).
Conclusion
Psuedo-science about melanin is rampant, no matter which side one is on. Both sides make ridiculous assertions and leaps of logic regarding melanin, and I find it very amusing that each group is talking about the same thing while attempting to argue the polar opposite of what the other is arguing. These misconceptions come from no understanding of physiology, to ideological biases, to delusions of ‘superiority’ to just plain ignorance overall. Afrocentrist fairy tales most probably are widening and already-wide science gap between blacks and whites. Of course, race doesn’t really have any bearing on whether or not you’ll believe something, though of course, black kids are more susceptible to believing the fantastical stories and non-understandings of physiology that come from their inner-city teachers who will then indoctrinate them to their ideology.
Correlations between skin pigmentation and neuromelanin are nonexistent. Further, there is no known physiological relationship between melatonin and skin color in humans. Therefore, the assertion that blacks have more melatonin due to their skin color and they then have this physical and mental superiority due to melanin has absolutely no scientific basis (even though those who push these types of theories have absolutely no understanding of the physiology of the hormone they are discussing). Racial pride ‘stories’ are harmful to science education; I don’t see March of the Titans being taught at schools (if I am in error let me know), but Afrocentric melanist theories are?
The most important thing to take note of here is the similarities between Rushton and Templer (2012) and melanists. They mirror each other so well, they are talking about the same exact hormone, but both groups have wildly different conclusions. Rushton and Templer (2012) were driven by the (wrong) hypothesis that testosterone caused aggression and crime and that since a whole slew of animals that had dark pigmentation were aggressive, therefore this should apply to humans too because “Evolution doesn’t stop at the neck”, as most people say. On the other side, we have melanists making wild, almost sci-fi like claims about the power and magic of this one hormone in black bodies and only black bodies. To believe something like that you’d have to be either ignorant or highly biased.
Melanism is clearly untenable, and Afrocentrists who push this ‘theory’ should take a few physiology classes and learn what this hormone does in the human body because they are woefully misinformed, reading books of pseudo-science.
The Weight Loss and Thermodynamics Fallacy
2000 words
Eat less and move more and you will lose weight. That’s the common mantra of everyone around the world because this is what has been repeated for decades. “The First Law of Thermodynamics states that energy can neither be created nor destroyed in an isolated system”. This Law is used in support of the CICO paradigm. But this kind of thinking does not make sense. The First Law only tells us that energy is conserved. That’s it. It says absolutely nothing about weight loss. Does anyone think that it’s weird that we’re given weight loss advice with physics (thermodynamics) and not advice for our physiology? This fallacy, what I term the CICO fallacy, then leads to the second fallacy: that a calorie is a calorie. The implication is this: the body does not discern between what type of macro you choose to ingest, it’s only worried about the amount of energy consumed. But, as I will show, this type of thinking does not work, either.
The First Law of Thermodynamics
The First Law states that energy can neither be created nor destroyed. A positive caloric balance must be associated with weight gain, but where the wrong conclusions come in is when people assume that the positive caloric balance is driving the weight gain. So if the First Law is interpreted correctly, then both conclusions—getting fat makes one consume more energy and consuming more energy makes one fat—are both valid hypotheses. The evidence and observations suggest that getting fat makes one consume more energy. (Jason Fung (2016: 33) writes: “Having studied a full year of thermodynamics in university, I can assure you that neither calories nor weight loss were mentioned even a single time.“)
Obesity researcher Jules Hirsch said to the New York Times:
There is an inflexible law of physics—energy taken in must exactly equal the number of calories leaving the system when fat storage is unchanged. Calories leave the system when food is used to fuel the body. To lower fat content—reduce obesity—one must reduce calories taken in, or increase activity, or both. This is true whether calories come from pumpkins or peanuts or pate de foie gras.
It’s this type of information that has caused the CICO paradigm to continue unabated. However, there are dissenting voices. People like Dr. Jason Fung, Gary Taubes, Zoe Harcombe, Nina Teicholz, Tim Noakes all go against the conventional wisdom regarding obesity and the cause for weight gain.
Another thing that is not taken into account is what occurs in the body when calories are reduced. One important thing to note is that the energy we consume and expend are not independent variables—they are dependent. Therefore, if we lower what we consume, what we expend will then lower as well. If you change one of them, the other will change too. For example, if you exercise more in an attempt to lose more weight you will eat more to compensate. If you eat less to lose more weight, your body’s metabolism will drop to match what the intake is. This is exactly what was seen in the Biggest Loser Study—shockingly lower RMRs in the contestants (see Fothergill et al, 2016). Biological systems are way more complex than to reduce it down to “eat less and move more=weight loss”, and that is easily shown.
Fliers and Maratos-Flier (2007: 74) write in Scientific American:
An animal whose food is suddenly restricted tends to reduce its energy expenditure both by being less active and by slowing energy use in cells, thereby limiting weight loss. It also experiences increased hunger so that once the restriction ends, it will eat more than its prior norm until the earlier weight is attained.
Take this example. Caloric excess in children is positively correlated with height increases. Though the caloric excess is not driving the height increases; they eat because they are growing.
The point that most people miss is the third storage system—fat storage. The three storage systems are kcal in/kcal out and fat storage. Insulin dictates fat storage, in the absence of insulin, the body cannot gain weight. Insulin shuttles fat into the adipocyte which is why insulin is fattening. That’s the point that CICO doesn’t work due to hormonal fluctuations. The most fattening hormone is insulin. The types of foods that elicit the highest insulin response are processed carbohydrates. Therefore, those are the most fattening foods. People who assume CICO state that a calorie is a calorie; that’s wrong.
Imagine a crowded room. The room is getting more crowded, and you ask me why the room is getting more crowded. I say ‘the room is more crowded because more people are entering it than leaving it.’ You say ‘duh, of course that’s true, but why is the room more crowded?’ Saying a room gets crowded because more people are entering than leaving it is redundant; saying that one gets fat because more calories are consumed than burned is redundant, it only says the same thing in two different ways so it is meaningless. Rooms that have more people enter them than leave them will become more crowded since there is no getting around the First Law, right?
Now take that same logic with obesity. Thermodynamics states that if we get fatter then more energy is entering our body than leaving it. Overeating means we’ve consumed more calories than we have expended. It’s tautological.
‘CICO could work’ but that is irrelevant, since what is assumed by the CICOers is that calories are calories; the assumption that once ingested, they go through the same metabolic pathways. This is false. The First Law says nothing about why we get fat. It is irrelevant to human physiology.
Taubes (2007: 293) writes:
Change in energy stores = Energy intake — Energy expenditure
[…]
The first law of thermodynamics dictates that weight gain—the increase in energy stored as fat and lean-tissue mass—will be accompanied by or associated with positive energy balance, but it does not say that it is caused by a positive energy balance—by “a plethora of calories,” as Russel Cecil and Robert Loeb’s 1951 Textbook of Medicine put it. There is no arrow of causality in the equation. It is equally possible, without violating this fundamental truth, for a change in energy stores, the left side of the above equation, to be the driving force in the cause and effect; some regulatory phenomenon could drive us to gai weight, which would in turn cause a positive energy balance—and thus overeating or sedentary behavior. Either way, the calories in will equal the calories out, as they must, but what is the cause in one cause is effect in the other.
And on pg 294:
The alternative hypothesis reverses the causality: we are driven to get fat by “primary metabolic or enzymatic effects,” as Hilde Bruch phrased it, and this fattening process induces the compensatory responses of overeating and/or physical inactivity. We eat more, move less, and have less energy to expend because we are metabolically or hormonally driven to get fat.
All the first law of thermodynamics tells us is that people can’t become more massive without taking in more energy than they expend since people who are heavier contain more energy than people who are lighter. That person has to consume more energy to accommodate said increasing mass. That person also cannot become lighter without expending more energy than they take in. That’s all the First Law tells us: energy is conserved. It says nothing about causation. The First Law literally only says that if something becomes more massive than more energy has to come in than leave. Nothing is said about cause and effect; it only tells us what has to happen if said thing does happen. That’s not causal information.
People only assume that the First Law has any relevance to obesity because of the ‘energy cannot be created nor destroyed’ part. But this shows no understanding of the Law. If you carefully read and understand it, you will see that it gives you absolutely no causal information. You can then reverse the commonly-held mantra—that eating more leads to obesity—to becoming obese leads one to eat more. It’s perfectly logical to reverse it and no Law is broken. People erroneously assume that the Laws of physics dictate weight gain and loss, but in complex metabolic systems, what is ingested is more important than how much is ingested (because we have hormones that let us know when to stop eating—which don’t get released while one eats carbohydrates).
The Second Law of Thermodynamics
The second weight loss fallacy is ‘a calorie is a calorie’, therefore, for weight loss, it doesn’t matter if a majority of my calories comes from fat, carbs or protein; the body will register the calories consumed and will regulate fat stores as dictated by the First Law (supposedly). The fallacy of invoking the First Law of thermodynamics ties directly into the fallacy of the Second Law of Thermodynamics—what the Second Law states is, that variation in metabolic pathways is to be expected, therefore, the mantra “a calorie is a calorie” violates the Second Law as a principle (Feinman and Fine, 2004, 2007).
A diet split of 55:30:15 CHO, fat, protein, yielded 1848 kcal. In fact, thermodynamics does not support the dictum that, all else being equal (i.e., two diets with the same amount of calories, but differing macro splits; one high-fat low carb the other high carb low-fat).
However, in 2004 Zoe Harcombe recalculated the figure from Feinman and Fein (2004) and found it to be wrong. The correct number ended up being 1825 kcal, not 1848 kcal, which strengthened Feinman and Fine’s (2004) point (Harcombe, 2004). She also writes:
I then repeated the calculations for a 10:30:60 high protein diet (keeping fat the same and swapping carbs out and protein in), and the calories available to the body dropped to 1,641. This is incredible. This means that two people can both eat 2000 calories a day and the high carbohydrate person is effectively getting nearly 200 calories more than the high protein person. Anyone still wonder why low-carbohydreate diets have a built in advantage?
So we can see that it’s ridiculous to ignore the thermic effect of food, seeing as it’s 20 percent for protein and 5 percent for CHO.
To put this into perspective, two people eating similar diets (but differing macro splits) only need to out-eat the other by 20 calories per day and that will be enough to gain more weight than the other person. Taubes (2011: 58) writes:
How many calories do we have to consume, but not expend, stashing them away in our fat tissue, to trsnsform ourselves, as many of us do, from lean twenty-five-year-olds to obese fifty-year-olds?
Twenty calories a day.
Twenty calories a day times the 365 days in a year comes to a little more than seven thousand calories stored as fat every year—two pounds of excess fat.
Multiply that by 10 and that’s twenty pounds gained in ten years—all from counting kcal wrong (Aamodt, 2016: 111-112). So with Harcomb’s (2004) example, the damage will be much worse in 10 years. This is all based on the assumption that ‘calories are calories’ which is false, as I have shown.
Conclusion
The CICO paradigm is wrong. Consumption and expenditure are not independent variables, they are dependent. So if you decrease one of them, the other will decrease as well. This is the fatal flaw in the CICO paradigm. The First Law always holds, yes, but it tells us absolutely nothing about obesity or human physiology and is therefore irrelevant. The Second Law is violated when one states that ‘a calorie is a calorie’, but this is demonstrably false. The Second Law states that variation in metabolic pathway efficiency is to be expected. Therefore stating that “a calorie is a calorie” violates the Second Law. This has further implications. Using Taubes’ example of 20 calories per day, if people truly believe the CICO mantra then people eating the same exact number of calories will have different weight gains if the skew of carbs to fat is higher in one than the other. Couple that with what insulin does in the body and this exacerbates the problem.
Stating that thermodynamics has anything to do with weight loss is clearly fallacious.
Behavior Genetics and the Fallacy of Nature vs Nurture
3250 words
People appeal to moderate to high heritability estimates as evidence that a trait is controlled by genes. They then assume that because something has a high heritability then that it must show something about causation. The fact of the matter is, they do not. Heritability estimates assume a false dichotomy of nature vs nurture; it assumes that we can neatly partition genetic from environmental effects. It assumes that the higher a trait’s heritability the more genes control said trait. These are all false. One of the main ways that heritability is estimated is by the CTM (classic twin method). This method, though, has a ton of assumptions poured into it—most importantly, the assumption that DZ and MZ fraternal twins experience roughly equal environments—the equal environments assumption (EEA). Heritability studies are useless for humans; twin studies bias estimates upwards with a whole host of assumptions.
I will show that i) heritability estimates are highly flawed (due to erroneous assumptions); ii) nature vs nurture cannot be separated (like behavior geneticists claim) and so their main tool (the heritability estimate) should be discontinued; iii) genetic reductionism is not a tenable model due to what we now know about how genes work. All three of these reasons are enough to discontinue heritability estimates. If the nature vs nurture debate rests on a fallacy, and this fallacy is used as a vehicle for heritability estimates, then they should be discontinued for humans and only be used for breeding animals where they can control the environment fully (Schonemann, 1997; Moore and Shenk, 2016).
Heritability, twin studies, and equal environments
Back in 2014-2015, there was a debate in the criminological literature that had implications for heritability studies as a whole. Burt and Simons (2014) stated that it was time to get rid of heritability studies. Barnes et al (2015) responded that this was “a de facto form of censorship” (pg 2). Joseph et al (2015) respond to these accusations, writing, “It was good science and not “censorship” when earlier scientists called for ending studies based on craniometry, phrenology, and physiognomy, and any contemporary criminologist calling for the use of astrological charts to predict whether certain people will commit violent crimes would be justifiably ridiculed.” The main thing here, in my opinion, is that heritability estimates are based on an oversimplified (and wrong) model of the gene. Partitioning variance assumes that you can partition how much a trait is influenced by “nature” or “nurture” which is a false dichotomy (Moore, 2002; Schneider, 2007; Moore and Shenk, 2016).
More importantly, no “genes have been found” (I know that’s everyone’s favorite thing to hear) for traits that supposedly have high heritabilities. On page 179 of his book (nook version), Misbehaving Science, Controversy and the Development of Behavior Genetics Panofsky (2014) writes:
Molecular genetics has been a major dissapointment, if not an outright failure, in behavior genetics. Scientists have made many bold claims about genes for behavioral traits or mental disorders only to later retract them or to have them not be replicated by other scientists. Further, the findings that have been confirmed, or not yet falsified, have been few, far between, and small in magnitude.
There seems to be a huge disconnect between heritability estimates gleaned from twin studies and what the actual molecular genetic evidence says. This is because the EEA—that fraternal MZ twins experience roughly similar environments compared to fraternal DZ twins—is false. Fraternal MZ twins end up experiencing more similar environments when compared with fraternal DZ twins. Though most researchers attempt to save face by stating that MZ twins “seek out” and “elicit” their own environments which then makes them more similar compared to DZ twins. However, this is circular logic. The conclusion (that twins experience more similar environments) is in the premise, and therefore it is an invalid argument due to the logical fallacy. (It should also be noted that identical twins’ genes are not identical.)
Heritability studies assume an outdated model of the gene. The flaw regarding heritability estimates is simple: they imply a false dichotomy of nature vs nurture, while also assuming that genes and environment are independent, while the contribution to complex behaviors can be precisely quantified (Charney, 2013). This is one of the most critical parts of the heritability debate. Prenatal environments of DZ twins “can be significantly more stressful than that of DZ twins, and hence a cause of greater stress-related phenotypic concordance, the equal environment assumption will not hold in relation to behavioral phenotypes potentially associated with prenatal stress” (Charney, 2012: 20). This also is cause for concern regarding studies of twins reared apart. While twins are reared apart to eliminate shared environmental confounds, it cannot eliminate perhaps the most important confound of all—the prenatal environment (Moore and Shenk, 2016).
One of the most-cited studies regarding twins reared apart is Bouchard (1990). Though there are a whole slew of problems with this study.
1) You have the huge confound of similar environments before birth.
2) Full details for the MISTRA have never been published, so we don’t know how ‘separated’ the twins were. Though Bouchard et al do say that they were separated between 0 to 48.7 months (table 1) so some pairs spent at least 4 years together. Some of the twins even had reunions and spent a lot of time together.
3) They’re not representative and twins who do sign up for this research are self-selecting. Ken Richardson says in his book (2017, pg 55): “Twins generally tend to be self-selecting in any twin study. They may have responded to advertisements placed by investigators or have been prompted to do so by friends or family, on the grounds that they are alike. Remember, at least some of them knew each other prior to the study. Jay Joseph has suggested that the twins who elected to participate in all twin studies are likely to be more similar to one another than twins who chose not to participate. This makes it difficult to claim that the results would apply to the general population.”
4) And the results aren’t fully reported. Richardson also states that (2017, pg 55) “… of two IQ tests administered in the MISTRA, results have been published for one but not the other. No explanation was given for that omission. Could it be they produced different results?” He even states that attempts to get the data, by researchers like Jay Joseph, have been denied. Why would you refuse to publish, or give to another researcher, your data when asked?
We don’t know the relevant environments, the children’s average age at testing is closer to the biological mother than adopted mother; the biological mother and child will have reduced self-esteem and be more vulnerable to difficult situations, and in this sense they share environments; and conscious or unconscious bias make adopted children different from other family members. Adoption agencies also attempt to put children into similar homes as the biological mother too.
Charney (2012: 25) brings up an important point: “For phenotypes of any degree of complexity, DNA does not contain a determinate genetic program (analogous to the digital code of a computer) from which we can predict phenotype. If DNA were the sole carrier of information relevant to phenotype formation, and contained a genetic program sufficiently determinate that solely by reading it we could predict phenotype, then humans (and all other organisms) would be largely lacking in phenotypic plasticity.” Moore and Shenk (2016) also state that “we inherit developmental resources, not traits.”
1 For twin studies to be valid DZ twins and MZ fraternal twins would have to experience roughly equal environments. 2 Fraternal MZ twins experience much more similar environments than DZ twins. 3 Therefore the EEA is false and no genetic interpretations can be drawn from the data.
Heritability estimates cannot detangle genes and environment, and therefore they should be discontinued or reinterpreted (Joseph et al, 2015). Burt and Simons (2014: 110) also conclude: “Rejecting heritability studies and the false nature–nurture dichotomy and gene-centric model on which they are grounded is a necessary step forward that will pave the way for a reconceptualization of the link between the biological and the social in shaping criminal propensities in ways that are consistent with postgenomic knowledge“. I disagree with Barnes et al (2015) when they say that ending heritability estimates are “a defacto form of censorship“, because if nature vs nurture is a false dichotomy and the gene-centric model that heritability estimates rely on is wrong, then we need to either discontinue or reinterpret the estimates, not saying that ‘this is how much nature contributes to X and this is how much nurture contributes to Y’. (See also Richardson and Norgate, 2005 for more arguments regarding the EEA.)
Sapolsky (2017: 219) writes:
Oh, that’s right, humans. Of all species, heritability scores in humans plummet the most when shifting from a controlled experimental setting to considering the species’ full range of habitats. Just consider how much the heritability score for wearing earrings, with its gender split, has declined since 1958.
Heritability flaws
High heritability estimates have been used as evidence for causation—that genes control a large part of the trait in question. This reasoning, however, is highly flawed. People confuse “heritable” with “inheritable” (Moore and Shenk, 2016). Heritability does not inform us what causes a trait, how much environment contributes to a trait, nor does it tell us the relative influence of genes on a trait. Moore and Shenk (2016) agree with Joseph et al (2015) and Burt and Simons (2014) that heritability studies need to end, but Moore and Shenk’s reasoning slightly differs: they say we should end estimates because people confuse “heritable” with “inheritable”. Likewise, Guo (2000: 299) concurs, writing “it can be argued that the term ‘heritability’, which carries a strong conviction or connotation of something ‘heritable’ in everyday sense, is no longer suitable for use in human genetics and its use should be discontinued.”
Some may say that if a trait turns out to be mildly heritable then we can say that genes have some effect, but we know that genes affect all traits so it seems kind of redundant to have a useless measure that assumes a false dichotomy and relies on an outdated, additive model of the gene.
Rose (2006), too, agrees that heritability estimates imply a false dichotomy of nature vs nurture onto biological systems:
Biological systems are complex, non-linear, and non-additive. Heritability estimates are attempts to impose a simplistic and reified dichotomy (nature/nurture) on non-dichotomous processes.
Likewise, Lewontin (2006) argues we should be analyzing and studying causes, not variance.
There are numerous hereditarian scientific fallacies which include: 1) trait heritability does not predict what would occur when environments/genes change; 2) they’re inaccurate since they don’t account for gene-environment covariation or interaction while also ignoring nonadditive effects on behavior and cognitive ability; 3) molecular genetics does not show evidence that we can partition environment from genetic factors; 4) it wouldn’t tell us which traits are ‘genetic’ or not; and 5) proposed evolutionary models of human divergence are not supported by these studies (since heritability in the present doesn’t speak to what traits were like thousands of years ago) (Bailey, 1997).
Bailey (1997) brings up important arguments against the use of heritability, and even discusses fallacious writing from Rushton on the matter:
Rushton (1995), for example, thinks that if observed differences among the
racial groups that he defines are higher for traits that have high heritability within the groups, the hypothesis of genetically caused differences among the groups is supported.
Bailey (1997) then goes on to discuss three lakes: Otter lake, Welcome lake, and Bark lake. Otter lake has very high primary production, while Bark lake has very little and Welcome lake is somewhere in between (you can see that ‘Otter’, ‘Bark’ and ‘Welcome’ lakes are analogies for ‘Orientals’, ‘Blacks’, and ‘Whites’ as said by Rushton). But there is variation within the lakes, there are high production pockets of water in Bark lake while there are low production pockets of water in Otter lake. All three lakes are visited and measurements are taken. Bailey (1997) states that his conclusion would be that they differ in how much light each receives. Bailey (1997: 131) writes:
If I substitute three groups of people for my lakes, IQ for primary production, and genes for light levels, the fallacy of the slippery scale, as applied to human behaviour genetics, becomes clear. Even if we are sure that there is a difference among groups of people in IQ, and we are sure that IQ has high heritability within
each of the groups (i.e. variation in IQ is largely caused by genetic variation), we can make no inference about the cause of differences in IQ among the groups. The differences might be caused by genetic differences or they might not, but the heritability studies within the groups can’t help us make that judgment.

(Genes don’t cause IQ scores—or behavior—but that’s for another day.)
Heritability estimates for, say, IQ, are higher than any other trait in the animal kingdom. Heritability estimates for animal traits are low—lower than the stratospheric heritability of IQ. For example, heritability estimates of the bodyweight of farm animals is about 30 percent, which is the same for egg and milk production. Body fat in pigs and wool on sheep has a heritability of about 50 percent. But these estimates pale in comparison to the heritability estimates of IQ: estimates have been as high as 80 percent (but Schonemann, 1997 states it’s 60 percent but it’s as high as 80-90 percent today); this heritability estimate for IQ “surpasses almost anything found in the animal kingdom” (Schonemann, 1997: 104).
This high heritability estimate for IQ, of course, comes to us from the highly flawed twin studies discussed above. The reason why farmers and botanists use heritability estimates is that they can perfectly control the environment, and therefore get accurate—or close enough to it—estimates that will help them in their breeding efforts. Conversely, for humans, environments cannot be perfectly controlled and it is, of course, unethical to rear twins, MZ and DZ, in a controlled environment. Proponents of the twin method may say “It doesn’t matter if it’s flawed, it still shows there is a genetic component to trait X!”. But as discussed by Moore and Shenk (2016), that’s irrelevant because genetic factors influence all of our characteristics.
Heritability and causation
In the final section, I will shortly discuss how people fallaciously assume that high heritability estimates imply that a trait is strongly influenced by genetic factors.
In his essay in the book Postgenomics: Perspectives on Biology After the Genome, sociologist Aaron Panofsky (2016: 167; nook version) writes:
Heritability estimates do not help identify particular genes or ascertain their functions in development or physiology, and thus, by this way of thinking, they yield no causal information.
This is important to note: to those who truly believe that heritability estimates tell us anything about causation, how could they, logically, give us causal information if genes that lead to trait variation are not identified (Richardson, 2012)?
Panofsky (2014: 102-103) writes:
Experimental evidence from plants and animals suggest that shapes of the curves cannot inferred in advance and rarely follow the smooth, nonintersecting pattern like in figure 3.2. Thus true causal interpretations of heritability are hopeless and must be abandoned. Behavior geneticists did not claim direct experimental evidence, but they thought these various indirect lines of evidence provided a reasonable set of assumptions that would enable them to interpret heritability scores causally—provided they offer apporopriate, reasonable qualifications.
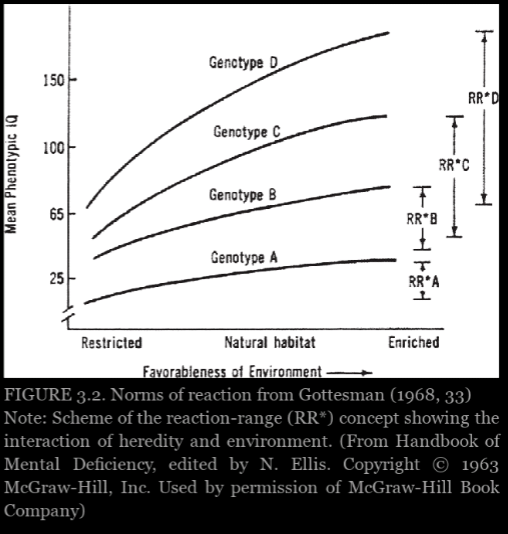
Graph from Panofsky (2014: 103)
Heritability estimates imply nothing about causation. It is about associations with variance, not identity and causes (Richardson, 2017: 69). A heritability of 0 does not mean that genes do not play a role in the development of form and function and phenotypic variation, it just means that, for whatever reason, there is little correlation between the two.
Scheneider (2007) writes (emphasis mine):
Heritability estimates apply only to groups, and are inherently inapplicable to individuals in any sense. And they do not imply causation. As Moore notes, all of these important limitations have been frequently ignored or minimized.
Reductionism
Heritability estimates imply nothing about causation. Behavior geneticists and others assume that heritability estimates will lead to ‘finding the genes’ that ’cause’ or are ‘associated with’ behavior. Their models are also, of course, extremely reductionist. It is then important to note that genes do not determine behavior. To quote Lerner and Overton (2017: 114):
Data presented in a 2016 special section of the journal Child Development indicate
that “some behaviors may be affected by only slight changes in DNA methylation,
while others may require a larger percent change in methylation; of course, the
effects are also likely bidirectional, with behavior impacting changes in methylation” [Lester et al., 2016, p. 31]. This point is key . It underscores the absurdity of genetic reductionist models: Genes do not determine behavior.
Methylation impacts behavior; behavior impacts methylation. It is the relations between methylation and behavior, not the genes acting as the “command center”, the “executive” of human behavior and development, that constitute the basic role of biology across the developmental course. This is the fatal flaw of reductionist models. Lastly, Lerner and Overton (2017: 145) write (emphasis mine):
That is, with the recent advances in understanding the role of epigenetics and recent research findings supporting this role, it should no longer be possible for any scientist to undertake the procedure of splitting of nature and nurture and, through reductionist procedures, come to conclusions that the one or the other plays a more important role in behavior and development.
[Richardson (2017: 129) also writes: “Note that this environmental source of [epigenetic] variation will appear in the behavioral geneticists twin-study as genetic variation: quite probably another way in which heritability estimates are distorted.”]
Reductionism in biology is fatally flawed. Reductionism, of course, has greatly increased our understanding of biology. However, it is time to move past the false dichotomy of nature vs nurture, and with that, move past heritability estimates since they prop up the fallacy of nature vs nurture. There is no way to separate the two since they are intertwined, but behavior geneticists would like you to believe that by studying twins raised apart will tell you anything about how ‘genetic’ or ‘environmental’ variation in a trait is in a population. Since heritability estimates are gleaned from the highly flawed studies of twins reared apart, a whole host of assumptions is poured in and these estimates are highly inflated, showing that genes influence a trait more than they supposedly do.
Twin studies, and along with it, heritability estimates, are useless for figuring out, and describing, trait variation in humans. The developmental system is more complex than the genetic reductionists (behavior geneticists) would like one to believe. The reductionist model has been heavily attacked in recent years (Regenmortal, 2004; Noble, 2008, 2012, 2015, 2016; Joyner, 2011, b; Joyner and Pederson, 2011).
Nature vs nurture has also shown to be a false dichotomy because the system develops in whichever environment it finds itself in (Oyama, 1985, 1999; 2000; Moore, 2002; Schneider, 2007)
Conclusion
Since the genetic reductionist model is wrong, along with heritability estimates (because of the nature/nurture fallacy), both should be discontinued. One of the main vehicles of these two models—twin studies—should also be discontinued. These fatal flaws of the behavior geneticists’ paradigm should be enough to discontinue these techniques in the study of human development and behavior. Heritability estimates give no causal information and they also use an outdated model of the gene; twin studies assume too many things for it to be a viable model in the discovering how traits manifest (most importantly, twin studies keep the nature/nurture fallacy alive and should be discontinued on that note only, in my opinion); and genetic reductionist models have been shown to be fatally flawed in recent years. We now have a better understanding of what a gene is today (Portin and Wilkins, 2017), and due to this, we should discontinue whatever implies the fallacy of nature vs nurture because it is irrelevant and a false dichotomy. That, alone, should be enough to discontinue twin studies and heritability estimates.
Race Differences in Penis Size Revisited: Is Rushton’s r/K Theory of Race Differences in Penis Length Confirmed?
2050 words
In 1985 JP Rushton, psychology professor at the University of Ontario, published a paper arguing that r/K selection theory (which he termed Differential K theory) explained and predicted outcomes of what he termed the three main races of humanity—Mongoloids, Negroids and Caucasoids (Rushton, 1985; 1997). Since Rushton’s three races differed on a whole suite of traits, he reasoned races that were more K-selected (Caucasoids and Mongoloids) had slower reproduction times, higher time preference, higher IQ etc in comparison to the more r-selected Negroids who had faster reproduction times, lower time preference, lower IQ etc (see Rushton, 1997 for a review; also see Van Lange, Rinderu, and Bushmen, 2017 for a replication of Rushton’s data not theory). Were Rushton’s assertions on race and penis size verified and do they lend credence to his Differential-K claims regarding human races?
Rushton’s so-called r/K continuum has a whole suite of traits on it. Ranging from brain size to speed of maturation to reaction time and IQ, these data points supposedly lend credence to Rushton’s Differential-K theory of human differences. Penis size is, of course, important for Rushton’s theory due to what he’s said about it in interviews.
Rushton’s main reasoning for penis size differences between race is “You can’t have both”, and that if you have a larger brain then you must have a smaller penis; if you have a smaller penis you must have a larger brain. He believed there was a “tradeoff” between brain size and penis size. In the book Darwin’s Athletes: How Sport Has Damaged Black America and Preserved the Myth of Race, Hoberman (1997: 312) quotes Rushton: “Even if you take something like athletic ability or sexuality—not to reinforce stereotypes or some such thing—but, you know, it’s a trade-off: more brain or more penis. You can’t have both.” This, though, is false. There is no type of evidence to imply that this so-called ‘trade-off’ exists. In my readings of Rushton’s work over the years, that’s always something I’ve wondered: was Rushton implying that large penises take more energy to have and therefore the trade-off exists due to this supposed relationship?
Andrew Joyce of the Occidental Observer published an article the other day in defense of Richard Lynn. Near the end of his article he writes:
Another tactic is to belittle an entire area of research by picking out a particularly counter-intuitive example that the public can be depended on to regard as ridiculous. A good example is J. Philippe Rushton’s claim, based on data he compiled for his classic Race, Evolution and Behavior, that average penis size varied between races in accord with the predictions of r/K theory. This claim was held up to ridicule by the likes of Richard Lewontin and other crusaders against race realism, and it is regularly presented in articles hostile to the race realist perspective. Richard Lynn’s response, as always, was to gather more data—from 113 populations. And unsurprisingly for those who keep up with this area of research, he found that indeed the data confirmedRushton’s original claim.
The claim was ridiculed because it was ridiculous. This paper by Lynn (2013) titled Rushton’s r-K life history theory of race differences in penis length and circumference examined in 113 populations is the paper that supposedly verifies Rushton’s theory regarding race differences in penis size, along with one of its correlates in Rushton’s theory (testosterone). Lynn (2013) proclaims that East Asians are the most K-evolved, then come Europeans, while Africans are the least K-evolved. This, then, is the cause of the supposed racial differences in penis size.
Lynn (2013) begins by briefly discussing Rushton’s ‘findings’ on racial differences in penis size while also giving an overview of Rushton’s debunked r/K selection theory. He then discusses some of Rushton’s studies (which I will describe briefly below) along with stories from antiquity of the supposed larger penis size of African males.
Our old friend testosterone also makes an appearance in this paper. Lynn (2013: 262) writes:
Testosterone is a determinant of aggression (Book, Starzyk, & Quinsey, 2001; Brooks & Reddon, 1996; Dabbs, 2000). Hence, a reduction of aggression and sexual competitiveness between men in the colder climates would have been achieved by a reduction of testosterone, entailing the race differences in testosterone (Negroids > Caucasoids > Mongoloids) that are given in Lynn (1990). The reduction of testosterone had the effect of reducing penis length, for which evidence is given by Widodsky and Greene (1940).
Phew, there’s a lot to unpack here. (I discuss Lynn 1990 in this article.) Testosterone does not determine aggression; see my most recent article on testosterone (aggression increases testosterone; testosterone does not increase aggression. Book, Starzyk and Quinsey, 2001 show a .14 correlation between testosterone and aggression, whereas Archer, Graham-Kevan, and Davies 2005 show the correlation is .08). This is just a correlation. Sapolsky (1997: 113) writes:
Okay, suppose you note a correlation between levels of aggression and levels of testosterone among these normal males. This could be because (a) testosterone elevates aggression; (b) aggression elevates testosterone secretion; (c) neither causes the other. There’s a huge bias to assume option a while b is the answer. Study after study has shown that when you examine testosterone when males are first placed together in the social group, testosterone levels predict nothing about who is going to be aggressive. The subsequent behavioral differences drive the hormonal changes, not the other way around.
Brooks and Reddon (1996) also only show relationships with testosterone and aggressive acts; they show no causation. This same relationship was noted by Dabbs (2000; another Lynn 2013 citation) in prisoners. More violent prisoners were seen to have higher testosterone, but there is a caveat here too: being aggressive stimulates testosterone production so of course they had higher levels of testosterone; this is not evidence for testosterone causing aggression.
Another problem with that paragraph quoted from Lynn (2013) is that it’s a just-so story. It’s an ad-hoc explanation. You notice something with data you have today and then you imagine a nice-sounding story to attempt to explain your data in an evolutionary context. Nice-sounding stories are cool and all and I’m sure everyone loves a nicely told story, but when it comes to evolutionary theory I’d like theories that can be independently verified of the data they’re trying to explain.
My last problem with that paragraph from Lynn (2013) is his final citation: he cites it as evidence that the reduction of testosterone affects penis length…..but his citation (Widodsky and Green, 1940) is a study on rats… While these studies can give us a wealth of information regarding our physiologic systems (at least showing us which types of avenues to pursue; see my previous article on myostatin), they don’t really mean anything for humans; especially this study on the application of testosterone to the penis of a rat. See, the fatal flaw in these assertions is this: would a, say, 5 percent difference in testosterone lead to a larger penis as if there is a dose-response relationship between testosterone and penis length? It doesn’t make any sense.
Lynn (2013), though, says that Rushton’s theory doesn’t propose that there is a direct causal relationship between “intelligence”‘ and penis length, but just that they co-evolved together, with testosterone reduction occurring when Homo sapiens migrated north out of Africa they needed to cooperate more so selection for lower levels of testosterone subsequently occurred which then shrunk the penises of Rushton’s Caucasian and Mongoloid races.
Lynn (2013) then discusses two “new datasets”, one of which is apparently in Donald Templer’s book Is Size Important (which is on my to-read list, so many books, so little time). Table 1 below is from Lynn reproducing Templer’s ‘work’ in his book.
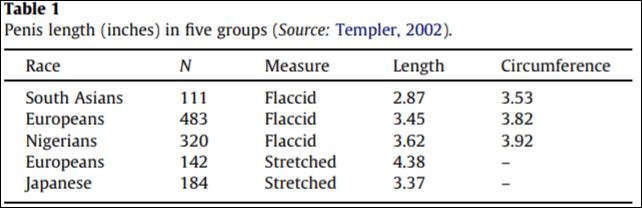
The second “dataset” is extremely dubious. Lynn (2013) attempts to dress it up, writing that “The information in this website has been collated from data obtained by research centres and reports worldwide.” Ethnicmuse has a good article on the pitfalls of Lynn’s (2013) article. (Also read Scott McGreal’s rebuttal.)
Rushton attempted to link race and penis size for 30 years. In a paper with Bogaert (Rushton and Bogaert, 1987), they attempt to show that blacks had larger penises than whites who h ad longer penises than Asians which then supposedly verified one dimension of Rushton’s theory. Rushton (1988) also discusses race differences in penis size, citing a previous paper by Rushton and Bogaert, where they use data from Alfred Kinsey, but this data is nonrepresentative and nonrandom (see Zuckermann and Brody, 1988 and Weizmann et al, 1990: 8).
Still others may attempt to use supposed differences in IGF-1 (insulin-like growth factor 1) as evidence that there is, at least, physiological evidence for the claim that black men have larger penises than white men, though I discussed that back in December of 2016 and found it strongly lacking.
Rushton (1997: 182) shows a table of racial differences in penis size which was supposedly collected by the WHO (World Health Organization). Though a closer look shows this is not true. Ethnicmuse writes:
ANALYSIS: The WHO did not study penis sizes. It relied on three separate studies, two of which were not peer-reviewed and the data was included as “Appendix III” (which should have alerted Rushton that this was not an original study). The first study references Africans in the US (not Africa!) and Europeans in the US (not Europe!), the second Europeans in Australia (not Europe!) and the third, Thais.
So it seems to be bullshit all the way down.
Ajmani et al (1985) showed that 385 healthy Nigerians had an average penile length of 3.21 inches (flaccid). Orakwe and Ebuh (2007) show that while Nigerians had longer penises than other ethnies tested, the only statistical difference was between them and Koreans. Though Veale et al (2014: 983) write that “There are no indications of differences in racial variability in our present study, e.g. the study from Nigeria was not a positive outlier.”
Lynn and Dutton have attempted to use androgen differentials between the races as evidence for racial differences in penis size (this is another attempt at a physiological argument to attempt to show the existence of racial differences in penis size). Edward Dutton attempted to revive the debate on racial differences in penis size during a 2015 presentation where he, again, showed that Negroids have higher levels of testosterone than Caucasoids who have higher levels of androgens than Mongoloids. These claims, though, have been rebutted by Scott McGreal who showed that populations differences in androgen levels are meaningless while they subsequently fail to validate Rushton and Lynn’s claims on racial differences in penis size.
Finally, it was reported the other day that condoms from China were too small in Zimbabwe, per Zimbabwe’s health minister. This led Kevin MacDonald to proclaim that this was “More corroboration of race differences in penis size which was part of the data Philippe Rushton used in his theory of r/K selection (along with brain size, maturation rates, IQ, etc.)” This isn’t “more corroboration” for Rushton’s long-dead theory; nor is this evidence that blacks have longer penises. I don’t understand why people make broad and sweeping generalizations. It’s one country in Africa that complained about smaller condoms from a country in East Asia, therefore this is more corroboration for Rushton’s r/K selection theory? The logic doesn’t follow.
Asians have small condoms. Those condoms go to Africa. They complain condoms from China are too small. Therefore Rushton’s r/K selection theory is corroborated. Flawed logic.
In sum, Lynn (2013) didn’t verify Rushton’s theory regarding racial differences in penis size and I find it even funnier that Lynn ends his article talking about “falsification’ stating that this aspect of Rushton’s theory has survived two attempts at falsification, therefore, it can be regarded as a “progressive research program“, though obviously, with the highly flawed “data” that was used, one cannot rationally make that statement. Supposed hormonal differences between the races do not cause penis size differences; even if blacks had levels of testosterone significantly higher than whites (the 19 percent that is claimed by Lynn and Rushton off of one highly flawed study in Ross et al, 1986) they still would not have longer penises.
The study of physical differences between populations is important, but sometimes, stereotypes do not tell you anything, especially in this case. Though in this instance, the claim that blacks have the longest penis lies on shaky ground, and with what evidence we do have for the claim, we cannot logically make the inference (especially not from Lynn’s (2013) flimsy data). Richard Lynn did not “confirm” anything with this paper; the only thing he “confirmed” are his own preconceived notions; he did not ‘prove’ what he set out to.
‘Double-Muscled’ Humans?
1800 words
I’ve been reading bodybuilding magazines for almost ten years. Good science articles on training and diet, but there was always one ad in the magazines that I always saw: the leg and calf of a neonate and then that same neonate at 7 months. The kid was brolic. Defined calves with absolutely no training. What was the cause? Well, he had a deletion on the myostatin gene— also called growth-differentiating factor 8, GDF-8. The ads in the magazines would try to get you to buy some shitty supplement that did not work, but the kid? The kid is real and he had a deletion on the gene that codes for a protein that myostatin. Myostatin restrains muscle growth, normally, which ensures that muscles don’t grow too large. Myo means muscle, while statin means heart. This can be a huge breakthrough regarding muscular dystrophy (Smith and Lin, 2013). Myostatin seems to have two roles: 1) regulating the number of muscle fibers formed in development and 2) to regulate the growth of muscle fibers postnatally.
When it is deleted, in cattle, it causes “double-muscle” cattle—cattle that have about 20 percent more muscle mass than cattle who don’t have the deletion (Grobert et al, 1997; Amthor et al, 2007). The cause is skeletal-muscle hyperplasia, which causes an increase in the number of muscle fibers, not only an increase in diameter. This is what causes these crazy-looking animals. Myostatin is coded by the MSTN gene. So they discovered that the gene caused double-muscled cattle. It should also be noted that while mice who lack myostatin are more muscular than average, they have impaired force generation (Amthor et al, 2007)

From Grobet et al, 1997; a double-muscle Belgian Blue homozygous for a deletion in the myostatin gene
The same thing is seen in mice—mice with a myostatin deletion are stronger and bigger (muscle) than mice without the myostatin deletion; myostatin, in adult mice, is expressed in all muscle tissue but more specifically in fast twitch muscle fibers (Whittemore et al, 2002). Se-Jin Lee is the one to discover the myostatin gene, and for his work he was elected the to the National Academy of Sciences in 2012 (Glass and Spiegelman, 2012). Mice that lack myostatin have, on average, double the muscle compared to mice who have myostatin. However, Lee (2007) proved that mice who lack myostatin and who overproduce follistatin (which is capable of blocking myostatin activity in muscle cells). Lee (2007) writes:
Moreover, the rank order of magnitude of these increases correlated with the rank order of expression levels of the transgene; in the highest-expressing line, Z116A [Z116a is one of Lee’s four transgenic mouse lines], muscle weights were increased by 57–81% in females and 87–116% in males compared to wild type mice. Hence, FLRG is capable of increasing muscle growth in a dose-dependent manner when expressed as a transgene in skeletal muscle.
So Lee (2007) discovered that the effect of FLRG is additive. He then attempted to determine whether or not the FLRG gene was truly causing increased muscle growth by blocking myostatin activity, so he examined the effect of combining the FLRG transgene with a knocked-out myostatin gene. He was not able to find this relationship in Z116A—i.e. being positive for the FLRG transgene and homozygous for myostatin—but he did discover that females from the Z166A strain were heterozygous for the myostatin deletion, having further increases in muscle weights combined with wild-type mice with ‘normal’ myostatin.
Most importantly, in two of the muscles that were examined (quadriceps and gastrocnemius) the observed increases were also greater than those seen in Mstn−/− mice lacking the transgene. Based on this finding, it appears that myostatin cannot be the sole target for FLRG in the transgenic mice and, therefore, that additional ligands must be capable of suppressing muscle growth in vivo.
Then Lee examined the effects of follistatin in MSTN null mice. He found that the presence of the F66 transgene in MSTN null mice, which caused another doubling in muscle. Lee had bred mice with quadruple muscle. Like FLRG, follistatin exerts its effects on other ligands, along with myostatin, so the effect of blocking still other ligands is also comparable to that loss-of-function from the myostatin.
So there are two important take-aways here with this landmark study: 1) the loss-of-function mutation on the Mstn gene exerts a maternal effect; muscle mass in the fetus is determined by the number of functional Mstn alleles (the offspring had higher muscle weights if the mother had fewer functioning Mstn alleles even if the offspring had the same genotype); and 2) Lee showed that other ligands worked with myostatin to control muscle growth. Both FLRG and follistatin can promote muscle growth when they are transgenes in skeletal muscle. So when he combined follistatin transgene and myostatin null mutation deletions, he had bred mice with qaudruple muscle.
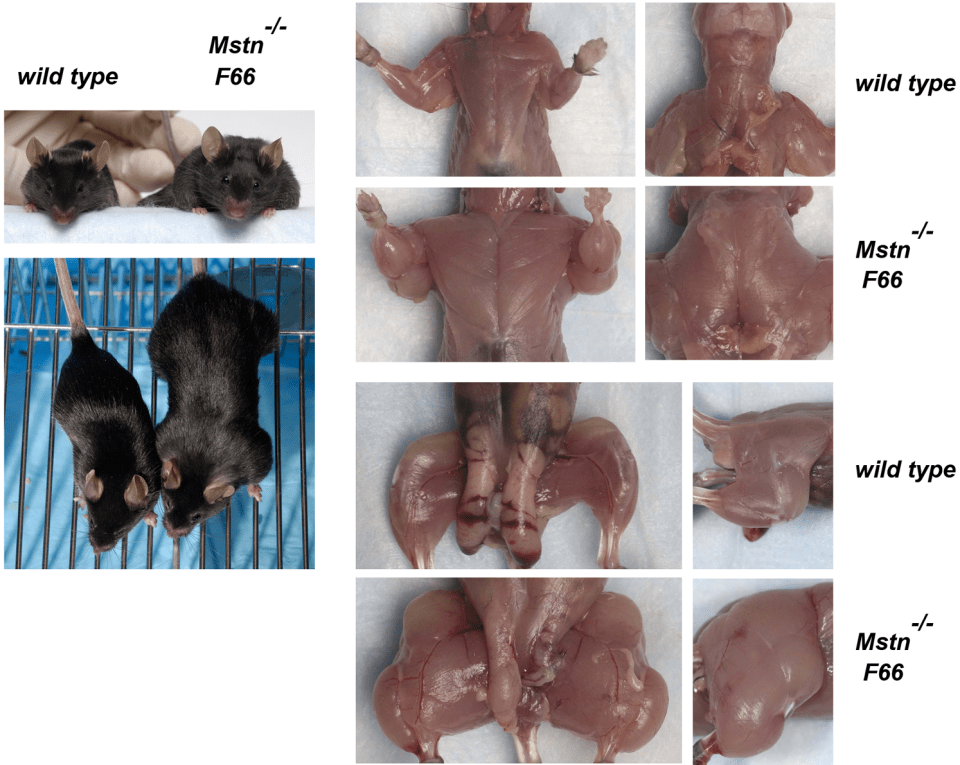
These mice are huge. And it’s only due simply to a loss-of-function mutation along with the myostatin-binding protein follistatin that causes mice with quadruple muscle. Myostatin regulates muscle growth. So if myostatin regulates muscle growth, then a deletion on the gene that codes for the protein that codes for myostatin to regulate muscle growth should causes increases in size and strength in animals with this null myostatin deletion.
In his 2014 book, David Epstein writes about how Lee attempted to find him subjects for human testing, so he put an ad in muscle magazines such as Muscle and Fitness and Muscular Development. Over 150 people answered his ad, but he had found no myostatin mutants.
This was until 2003, when he got a phone call of a babe who was born with bulging muscles, in Germany. He had mutations on both of his myostatin genes, therefore he had no myostatin in his blood. This baby’s mother (called “Superbaby”) had one normal myostatin gene and one mutant so she had more myostatin than her son but less than the general population. She is the only adult with a known myostatin deletion, and she just so happens to be a professional sprinter.
Before I discuss Superbaby, I need to discuss myostatin and its role in development. Myostatin plays the same role in birds, cattle, mice, humans, etc. Muscle is costly, energetically speaking, and if one is too muscular they may not be able to find enough food to sustain their higher-than-average muscle mass, so myostatin is kind of like the body’s ‘fail-safe’ to prevent one’s muscles from becoming too big. Of course larger muscles require more calories—and of course protein for muscle-building—and so, it wouldn’t make sense, for instance, for our ancestors to have huge bulging muscles since they ate intermittently. So myostatin helps us stay smaller than we would be than if we had the null mutation.
One of the incredible things about Superbaby is that he had no heart problems, although doctors were worried that he would (like his heart growing out of control), but him nor his mother have reported any problems. Epstein (2014: 105) writes:
But the facts that the one boy with two of the rare myostatin gene variants has exceptional strength, and that his mother has exceptional speed, are no coincidence. Superbaby and his mother fall precisely in line with whippets.
Epstein describes how two whippets, one with one copy of the myostatin gene, have four puppies and how the mutation would go to the offspring (kind of like a Punnet square):
If two sprinter whippets—dogs that each have one copy of the myostatin mutation—have four puppies, this is the likely scenario: one puppy will have zero copies of the mutation and be normal; two puppies will have one copy of the mutation, like Superbaby’s mother, and be sprinters; the fourth pippy will have two copies of the mutation, like Superbaby, which make for a double-muscled “bully” whippet.
Schuelke et al’s (2004) case report is one of the first-known cases of the myostatin mutation in humans. The pregnancy was normal, and when he was born, Superbaby had protruding calves (see Fig. 1: a below; left is as a 6-day-old neonate and the right is 7 months), along with his upper arms too. The ultrasonograms of his muscles were also different from controls as was the morphometric analysis (Fig. 1 b and c respectively). All around, Superbaby was normal; but by age 3 he still had increased muscle mass/strength and could even hold 3 kg dumbells in suspension, horizontally with his arms extended. He had some strong family members, one was a construction worker who was able to unload curbstones by hand, while the mother “appeared muscular” but not as muscular as her son (see Fig. 1 d).
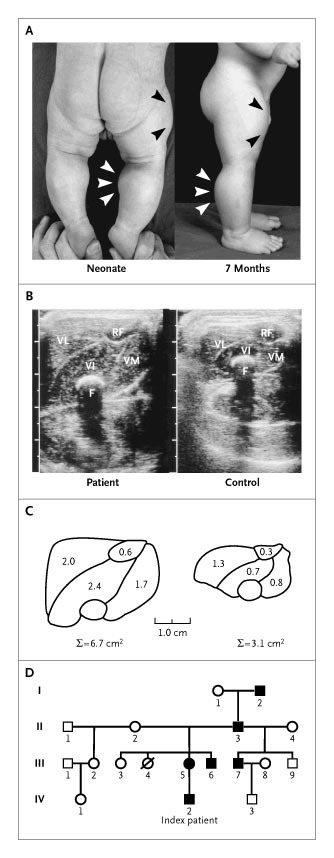
Myostatin is also expressed in the heart, and since Superbaby had a loss-of-function mutation on his myostatin gene, he was monitored for cardiomyopathy but he may have been too young to detect any defects. So Schuelke et al’s (2004) obvious conclusion was that a loss-of-function on the myostatin gene could increase muscle bulk and strength and be good therapy for people with a muscle-wasting disease.
One deletion in the MSTN gene can cause myostatin-related hypertrophy. One mutation disrupts how the gene that codes for the protein to make MSTN and therefore muscle cells. So when this occurs the cells make little to no functional myostatin. When one protein is lost, it leads to an overgrowth of muscle cells, with no other apparent medical problems (which is also seen in Superbaby and his mother).
If that were my son, I’d be a proud father. My baby coming out of the womb already jacked and strong? He’d immediately be in the gym as soon as he was able to and I would attempt to mold him into a champion bodybuilder/powerlifter; I’m not sure if the mutation that Superbaby has would truly matter at the elite level in the IFBB, but it would matter in the amateur level. Superbaby—and all of the other loss-of-function myostatin animal mutants—have paved the way for new forms of gene therapy for humans who have a muscle-wasting disease. Another American boy also had the same mutation as Superbaby. (Though the cause is different in this child, his body produces a normal level of myostatin, a defect in his myostatin receptors is thought to prevent his muscle cells from responding to myostatin, and since he’s bigger and stronger than children his age, this is a sensible hypothesis.)
With these loss-of-function mutants along with other transgenes, we can understand how and why muscles atrophy and grow, and we can help people with serious disease. Superbaby is not 14 years old, and while I am unable to find any new information on Superbaby (I will write something else on this if and when I do), it’s clear that a loss-of-function on the myostatin gene causes higher amounts of muscle mass and strength when people with the mutation are compared to people without the mutation. I’d personally line up turn off my myostatin gene, so I can get double-muscled and if there is any gene therapy for follistatin, I’d get that, too, in order to become quadruple-muscled.
