1800 words
I’ve been reading bodybuilding magazines for almost ten years. Good science articles on training and diet, but there was always one ad in the magazines that I always saw: the leg and calf of a neonate and then that same neonate at 7 months. The kid was brolic. Defined calves with absolutely no training. What was the cause? Well, he had a deletion on the myostatin gene— also called growth-differentiating factor 8, GDF-8. The ads in the magazines would try to get you to buy some shitty supplement that did not work, but the kid? The kid is real and he had a deletion on the gene that codes for a protein that myostatin. Myostatin restrains muscle growth, normally, which ensures that muscles don’t grow too large. Myo means muscle, while statin means heart. This can be a huge breakthrough regarding muscular dystrophy (Smith and Lin, 2013). Myostatin seems to have two roles: 1) regulating the number of muscle fibers formed in development and 2) to regulate the growth of muscle fibers postnatally.
When it is deleted, in cattle, it causes “double-muscle” cattle—cattle that have about 20 percent more muscle mass than cattle who don’t have the deletion (Grobert et al, 1997; Amthor et al, 2007). The cause is skeletal-muscle hyperplasia, which causes an increase in the number of muscle fibers, not only an increase in diameter. This is what causes these crazy-looking animals. Myostatin is coded by the MSTN gene. So they discovered that the gene caused double-muscled cattle. It should also be noted that while mice who lack myostatin are more muscular than average, they have impaired force generation (Amthor et al, 2007)

From Grobet et al, 1997; a double-muscle Belgian Blue homozygous for a deletion in the myostatin gene
The same thing is seen in mice—mice with a myostatin deletion are stronger and bigger (muscle) than mice without the myostatin deletion; myostatin, in adult mice, is expressed in all muscle tissue but more specifically in fast twitch muscle fibers (Whittemore et al, 2002). Se-Jin Lee is the one to discover the myostatin gene, and for his work he was elected the to the National Academy of Sciences in 2012 (Glass and Spiegelman, 2012). Mice that lack myostatin have, on average, double the muscle compared to mice who have myostatin. However, Lee (2007) proved that mice who lack myostatin and who overproduce follistatin (which is capable of blocking myostatin activity in muscle cells). Lee (2007) writes:
Moreover, the rank order of magnitude of these increases correlated with the rank order of expression levels of the transgene; in the highest-expressing line, Z116A [Z116a is one of Lee’s four transgenic mouse lines], muscle weights were increased by 57–81% in females and 87–116% in males compared to wild type mice. Hence, FLRG is capable of increasing muscle growth in a dose-dependent manner when expressed as a transgene in skeletal muscle.
So Lee (2007) discovered that the effect of FLRG is additive. He then attempted to determine whether or not the FLRG gene was truly causing increased muscle growth by blocking myostatin activity, so he examined the effect of combining the FLRG transgene with a knocked-out myostatin gene. He was not able to find this relationship in Z116A—i.e. being positive for the FLRG transgene and homozygous for myostatin—but he did discover that females from the Z166A strain were heterozygous for the myostatin deletion, having further increases in muscle weights combined with wild-type mice with ‘normal’ myostatin.
Most importantly, in two of the muscles that were examined (quadriceps and gastrocnemius) the observed increases were also greater than those seen in Mstn−/− mice lacking the transgene. Based on this finding, it appears that myostatin cannot be the sole target for FLRG in the transgenic mice and, therefore, that additional ligands must be capable of suppressing muscle growth in vivo.
Then Lee examined the effects of follistatin in MSTN null mice. He found that the presence of the F66 transgene in MSTN null mice, which caused another doubling in muscle. Lee had bred mice with quadruple muscle. Like FLRG, follistatin exerts its effects on other ligands, along with myostatin, so the effect of blocking still other ligands is also comparable to that loss-of-function from the myostatin.
So there are two important take-aways here with this landmark study: 1) the loss-of-function mutation on the Mstn gene exerts a maternal effect; muscle mass in the fetus is determined by the number of functional Mstn alleles (the offspring had higher muscle weights if the mother had fewer functioning Mstn alleles even if the offspring had the same genotype); and 2) Lee showed that other ligands worked with myostatin to control muscle growth. Both FLRG and follistatin can promote muscle growth when they are transgenes in skeletal muscle. So when he combined follistatin transgene and myostatin null mutation deletions, he had bred mice with qaudruple muscle.
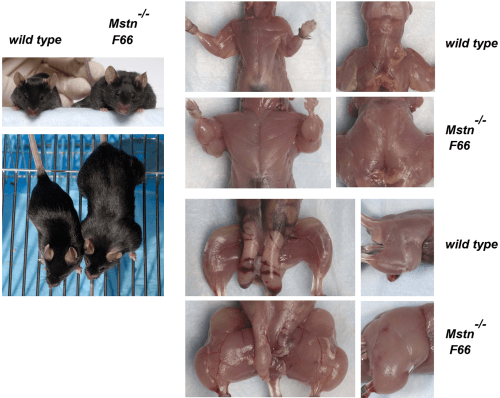
These mice are huge. And it’s only due simply to a loss-of-function mutation along with the myostatin-binding protein follistatin that causes mice with quadruple muscle. Myostatin regulates muscle growth. So if myostatin regulates muscle growth, then a deletion on the gene that codes for the protein that codes for myostatin to regulate muscle growth should causes increases in size and strength in animals with this null myostatin deletion.
In his 2014 book, David Epstein writes about how Lee attempted to find him subjects for human testing, so he put an ad in muscle magazines such as Muscle and Fitness and Muscular Development. Over 150 people answered his ad, but he had found no myostatin mutants.
This was until 2003, when he got a phone call of a babe who was born with bulging muscles, in Germany. He had mutations on both of his myostatin genes, therefore he had no myostatin in his blood. This baby’s mother (called “Superbaby”) had one normal myostatin gene and one mutant so she had more myostatin than her son but less than the general population. She is the only adult with a known myostatin deletion, and she just so happens to be a professional sprinter.
Before I discuss Superbaby, I need to discuss myostatin and its role in development. Myostatin plays the same role in birds, cattle, mice, humans, etc. Muscle is costly, energetically speaking, and if one is too muscular they may not be able to find enough food to sustain their higher-than-average muscle mass, so myostatin is kind of like the body’s ‘fail-safe’ to prevent one’s muscles from becoming too big. Of course larger muscles require more calories—and of course protein for muscle-building—and so, it wouldn’t make sense, for instance, for our ancestors to have huge bulging muscles since they ate intermittently. So myostatin helps us stay smaller than we would be than if we had the null mutation.
One of the incredible things about Superbaby is that he had no heart problems, although doctors were worried that he would (like his heart growing out of control), but him nor his mother have reported any problems. Epstein (2014: 105) writes:
But the facts that the one boy with two of the rare myostatin gene variants has exceptional strength, and that his mother has exceptional speed, are no coincidence. Superbaby and his mother fall precisely in line with whippets.
Epstein describes how two whippets, one with one copy of the myostatin gene, have four puppies and how the mutation would go to the offspring (kind of like a Punnet square):
If two sprinter whippets—dogs that each have one copy of the myostatin mutation—have four puppies, this is the likely scenario: one puppy will have zero copies of the mutation and be normal; two puppies will have one copy of the mutation, like Superbaby’s mother, and be sprinters; the fourth pippy will have two copies of the mutation, like Superbaby, which make for a double-muscled “bully” whippet.
Schuelke et al’s (2004) case report is one of the first-known cases of the myostatin mutation in humans. The pregnancy was normal, and when he was born, Superbaby had protruding calves (see Fig. 1: a below; left is as a 6-day-old neonate and the right is 7 months), along with his upper arms too. The ultrasonograms of his muscles were also different from controls as was the morphometric analysis (Fig. 1 b and c respectively). All around, Superbaby was normal; but by age 3 he still had increased muscle mass/strength and could even hold 3 kg dumbells in suspension, horizontally with his arms extended. He had some strong family members, one was a construction worker who was able to unload curbstones by hand, while the mother “appeared muscular” but not as muscular as her son (see Fig. 1 d).
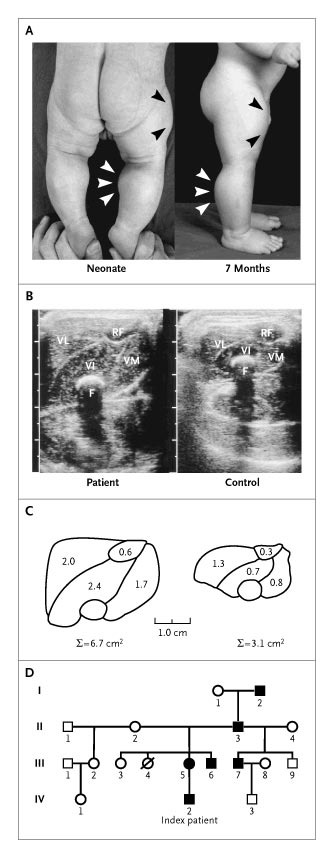
Myostatin is also expressed in the heart, and since Superbaby had a loss-of-function mutation on his myostatin gene, he was monitored for cardiomyopathy but he may have been too young to detect any defects. So Schuelke et al’s (2004) obvious conclusion was that a loss-of-function on the myostatin gene could increase muscle bulk and strength and be good therapy for people with a muscle-wasting disease.
One deletion in the MSTN gene can cause myostatin-related hypertrophy. One mutation disrupts how the gene that codes for the protein to make MSTN and therefore muscle cells. So when this occurs the cells make little to no functional myostatin. When one protein is lost, it leads to an overgrowth of muscle cells, with no other apparent medical problems (which is also seen in Superbaby and his mother).
If that were my son, I’d be a proud father. My baby coming out of the womb already jacked and strong? He’d immediately be in the gym as soon as he was able to and I would attempt to mold him into a champion bodybuilder/powerlifter; I’m not sure if the mutation that Superbaby has would truly matter at the elite level in the IFBB, but it would matter in the amateur level. Superbaby—and all of the other loss-of-function myostatin animal mutants—have paved the way for new forms of gene therapy for humans who have a muscle-wasting disease. Another American boy also had the same mutation as Superbaby. (Though the cause is different in this child, his body produces a normal level of myostatin, a defect in his myostatin receptors is thought to prevent his muscle cells from responding to myostatin, and since he’s bigger and stronger than children his age, this is a sensible hypothesis.)
With these loss-of-function mutants along with other transgenes, we can understand how and why muscles atrophy and grow, and we can help people with serious disease. Superbaby is not 14 years old, and while I am unable to find any new information on Superbaby (I will write something else on this if and when I do), it’s clear that a loss-of-function on the myostatin gene causes higher amounts of muscle mass and strength when people with the mutation are compared to people without the mutation. I’d personally line up turn off my myostatin gene, so I can get double-muscled and if there is any gene therapy for follistatin, I’d get that, too, in order to become quadruple-muscled.

Blah, just reviewed some of your articles… I think the Culture whiz is a lot closer to reality than you e.g. http://culturewhiz.org/forum/topic/human-brain-not-intelligence Seriously, is you Iphone intelligent? He nails that. He also explains the mental illness/high IQ connection http://culturewhiz.org/forum/topic/extreme-intelligence-mental-illness-related-autism I spent something like 25+ years of my life with a brain altered by a virus with secondary factors resulting in a higher rate of memory encoding than most humans, I have a heightened immune system because of the way the brain and immune system are linked (which doctors don’t even know about!! None of your articles take into acct that or bacterial fungus, etc http://culturewhiz.org/forum/topic/summary-most-advanced-medical-research
LikeLike
Good stuff there. I largely agree. I can see he has read Noble’s work too.
LikeLike
Thought you might be interested in this recent Instagram post by former WSM Eddie Hall, claiming that he had a mutation on the MSTN gene : https://www.instagram.com/p/Boeq7xRg9pO/?utm_source=ig_share_sheet&igshid=sc386mm26i93
LikeLike