Home » Nutrition
Category Archives: Nutrition
Differences in Body Composition Between Asians and Whites
1700 words
Black-white differences get talked about more than Asian-white differences. (For the purposes of this article, “Asian” refers to Koreans, Chinese, Japanese and Filipinos whereas “white” refers to those of European descent.) One interesting racial difference is that of body fatness between ethnies/races. Blacks have thinner skin folds and lower percent body fat than whites at the same height/BMI, and Asians have higher body fat and larger skinfolds than do whites. The interesting thing about this Asian-white difference is the fact that, at the same BMI, Asians have more upper body fat (trunk) than whites. The interesting thing is that there are two good studies, looking at these types of differences between Asians and whites (one study looking at the aforementioned “Asians” I previously identified and whites in the NYC area and another comparing whites and Chinese living in China.)
Wang et al (1994) studied 687 healthy volunteers (445 whites and 242 Asians, ranging from 18-94 years of age with BMIs in the range of 15-38). They defined ethnicity as the birthplace of one’s grandparents. The “Asian” category included 225 Chinese, 9 Japanese, 6 Koreans and 2 Filipinos; 97 percent of this sample was born in Asia. Then, after an overnight fast to better assess body fat differences and skinfold measures, they were weighed and measured, with their back, butt and feet firmly against the wall.
They measured skinfold thickness at the midarm for the average of the triceps and biceps, trunk thickness was the average circumference of the chest, subscapular, umbilicus, abdomen, and suprailiac. The circumference of the arm was measured at the midarm, while the circumference of the trunk was the average circumference of the upper chest, waist, iliac crest, and chest.
Wang et al (1994) found that whites were significantly taller, heavier and had higher BMIs than Asians. Wang et al (1994: 25) write:
For lean and normal BMIs, Asians were fatter than whites in both sexes, but the differences in estimated fat% between whites and Asians varied by BMI in different directions for males and females: fat% increased with BMI for males but decreased with BMI for females.
Whites were had significantly larger circumference in the measured appendages compared to Asians, while in Asian and white females, the circumference of the arms and waist were not different but other circumferences showed a greater difference, favoring whites. Asians had significantly higher levels of subcutaneous trunk fat (upper body fat) than whites, while white females had more lower (thigh) body fat than Asians. In both sexes, Asians had thicker bicep, subscapular, abdomen, and suprailiac skinfolds than whites, in both sexes. White women had higher levels of subcutaneous fat in their thighs. The only difference between white and Asian males in regard to skinfold area was the thigh, with whites having larger thighs, but were similar at the midarm and trunk. Asian men had a larger trunk skinfold area whereas whites had a larger thigh skinfold area while arm fatness did not differ between the races. Women in both races had larger skinfold areas except in the trunk; for whites, there were no differences between the sexes. In both sexes, Asians had higher values in subcutaneous fat (at the midarm, trunk, and midthigh), but white women had a higher value in the thigh than Asian women.
Wang et al (1994) show that there are significant differences in body fatness at different sites of the body, and so, since most (if not all) BMI equations are based on white populations, then, these equations will not work for Asians and will result in substantial error.
Wang et al (2011) studied differences in body composition between Chinese and white males living in the Shenzhen, Guangdong Province, China. They studied 115 Chinese and 114 white males. In this sample, Chinese males were younger, shorter, had a lower body weight and lower BMI than the white sample. Whites had higher fat mass, fat-free mass and bioelectrical impedance (which assess body composition, which measures lean mas in relation to fat mass; but these can be skewed by how much water one has or has not drunk, and so the DXA scan and hydrostatic weighing are, in my opinion, superior assessors). After adjustment for age and BMI, the percentage of fat mass in the trunk and arm was higher in Chinese than white males. Further, Chinese men had higher diastolic blood pressure (DBP), fasting glucose (FG) and triglycerides (TG), while whites had higher fasting total plasma cholesterol (TC) and high-density lipoprotein (HDL). The only statistically significant differences were between FG and HDL. Even after adjustment, Chinese men had 3.0 mmHg higher DBP than whites.
Chinese men had higher percent body fat than whites and more fat stored around their trunks than whites at the same BMI. Chinese men had higher fasting glucose levels (a risk-factor for obesity) but lower HDL levels at the same BMI as whites. Wang et al (2011) write:
In addition, comparing the two nationally representative studies, NHANES III [32] and China National Nutrition and Health Survey 2002 (CNNHS 2002) [33], Chinese men held a relatively 15.0% lower mean value of BMI than that for American white men. While comparison results from two large-scale epidemiological studies, the Shanghai Diabetes Studies (SHDS) [34] and the NHANES III [35], show that the mean value of PBF for American men is relatively 7.4% higher than that for Chinese men. The relative difference of PBF between American and Chinese males is much less than the difference of BMI, implying that the PBF among American men should be lower than that of Chinese men with the same BMI level.
What this implies is that the proportion of overweight/obese Chinese men are severely underestimated since, as noted earlier, most—if not all—BMI equations are created using strictly white populations. This study also provides more evidence that Chinese men had more central (trunk) adiposity than whites (Britons, in this study; Eston, Evans, and Fu, 1994). Central adiposity and risk for type II diabetes and cardiovascular disease is heightened in those of Chinese descent (Weng et al, 2006). It should also be noted that, in a sample of 129 Pacific Islanders, 120 Asians, 91 Maoris, and 91 Europeans aged 12-91, the relationship between bioelectrical impedance analysis (BIA) is ethnicity-dependent, due to the fact the equations developed for fatness estimation using BIA were more accurate than what was recommended by the manufacturer (Sluyter et al, 2010). Cheng (2011) showed that central adiposity was more predictive of cardiovascular diseases in the Chinese population than was BMI, while Hu et al (2007) showed that central obesity was more related to diabetes mellitus and impaired fasting glucose than to overall obesity in the Chinese population.
So, clearly, obesity-related factors appear at lower BMIs for Asians than Europeans (e.g., Huxley et al, 2008). Pan et al (2004) showed that for most BMI values, incidences of hypertension, diabetes, and hyperuricemia were higher in the Taiwanese sample than in the white and black samples. As BMI got higher, the risk for hypertriglyceridemia and hypertension increased. They showed that BMIs of 22.6, 26, and 27.5 were the cutoffs for the best predictabilty in regard to negative and positive variables for Taiwanese, white and black men, respectively. Pan et al (2004: 31) write:
For BMIs 27, 85% of Taiwanese, 66% of whites, and 55% of blacks had at least one of the studied comorbidities. However, a cutoff close to the median of the studied population was often found by maximizing sensitivity and specificity. Reducing BMI from 25 to 25 in persons in the United States could eliminate 13% of the obesity comorbidity studied. The corresponding cutoff in Taiwan is slightly 24.
Pan et al (2004) conclude that, for Taiwanese (Asians) in their study, they should have a lower BMI cutoff than whites and blacks, though it is tough to ascertain where that cutoff would be.
Bell, Adair, and Popkin (2002) show that “at BMI levels less than 25, prevalence difference figures suggested a stronger association between BMI and hypertension in Chinese men and women but not in Filipino women, compared with non-Hispanic Whites” while “[n]on-Hispanic Blacks and Filipino women had a higher prevalence of hypertension at every level of BMI compared with non-Hispanic Whites and Mexican Americans.”
Since Asians have a higher risk of hypertension than whites after controlling for BMI, this indicates that the effects of obesity are not as important as other factors, be they genetic or environmental (or both, which it obviously is). The higher incidence of obesity-related risk-factors in Asian populations with lower BMIs has been attributed to GxE interactions, which, of course, have been intensified with the introduction of the Western Diet (AKA the SAD [Standard American Diet] diet). This can be most notably seen with the explosion of childhood obesity in China, with the number of obese people in China surpassing the US recently, while China is on its way to have the most obese children in the world. The surging obesity epidemic in China is due to increasingly similar lifestyles to what we have (sedentary populations; highly processed, high fat, high carbohydrate foodstuff).
So since the findings in the reviewed studies suggest that, at a lower BMI, Asians are more susceptible to obesity-related risk-factors, and so, BMI standards must be lowered for Asian populations, which would be BMI 24 for overweight and BMI 27 for obese, which was recommended by the Chinese Ministry of Health (Wang et al, 2010). Cheung et al (2018) show that diet quality is inversely associated with obesity in Chinese adults who have type II diabetes.
In conclusion, Asians at the same BMI have higher body fat percentage than whites, and they also have more obesity-related risk-factors than whites at a lower BMI (Pan et al, 2004; WHO expert consultation, 2004; Wang et al, 2010; Hsu et al, 2015), which implies that they need differing BMI scales, just as blacks need different scales in comparison with whites. Here is a good example of two people with the same BMI (22.3) but different DXA results:

This, of course, shows the strong limitations of the use of the same BMI standards calculated in one ethny and used for another. So, just like at the same BMI blacks have lower body fat and thinner skinfolds than whites (Vickery, Cureton, and Collins, 1988; Wagner and Heyward, 2000; Flegal et al, 2010), at the same BMI as whites, Asians have higher body fat and thicker skinfolds (Wang et al, 1994; WHO expert consultation, 2004; Wang et al, 2011).
What Is the “Human Diet”?
3000 words
Is there one (or, one with slight modifications) diet that all humans should be eating? I’m skeptical of such claims. Though both vegans (one who does not eat or use animal products) and carnivores (one who eats only animal products), in my opinion, have some warped views on diet and human evolution. Both are extreme views; both have wrong ideas about diet throughout our evolution; both get some things right. Though, both are extreme views with little to no support. While it is hard to pinpoint what the “human diet” is, clearly, there were certain things that we ate through our evolutionary niches in our ancestral Africa that we “should” be eating today (in good quantities).
Although it is difficult to reconstruct the diet of early hominids due to lack of specimens (Luca, Perry, and Rienzo, 2014), by studying the eating behavior of our closest evolutionary relatives—the chimpanzees—we can get an idea of what our LCA ate and its eating behavior (Ulijaszek, Mann, and Elton, 2013). Humans have been throughout most every niche we could possibly been in and, therefore, have come across the most common foods in each ecology. If animal A is in ecosystem E with foods X, Y, and Z, then animal A eats foods X, Y, and Z, since animals consume what is in their ecosystem. Knowing this much, the niches our ancestors lived in in the past had to have a mix of both game and plants, therefore that was our diet (in differing amounts, obviously). But it is more complicated than that.
So, knowing this, according to Ulijaszek, Mann, and Elton, (2013: 35) “Mammalian comparisons may be more useful than ‘Stone Age’ perspectives, as many of the attributes of hominin diets and the behaviour associated with obtaining them were probably established well before the Pleistocene, the time stone agers were around (Foley 1995; Ulijaszek 2002; Elton 2008a).” Humans eat monocots (various flowering plants with one seed), which is not common our order. The advent of farming was us “expanding our dietary niche”, which began “the widespread adoption of agriculture [which] is an obvious point of transition to a ‘monocot world’” (Ulijaszek, Mann, and Elton, 2013). Although these foodstuffs dominate our diet, there is seasonality in what types of those foods we consume.
So since humans tend to not pick at things to eat, but have discrete meals (it is worth noting that one should have “three square meals a day” is a myth; see Mattson et al, 2014), we need to eat a lot in the times we do eat. Therefore, since we are large-bodied primates and our energy needs are much higher (due to our large brains that consume 20 percent of our daily caloric consumption), we need higher quality energy. The overall quality and energy density of our diets are due to meat-eating—which folivorous/frugivorous primates do not consume. We have a shorter gut tract which is “often attributed to the greater reliance of faunivory in humans“, though “humans are not confined to ‘browse’ vegetation … and make extensive use of grasses and their animal consumers” (Ulijaszek, Mann, and Elton, 2013: 58). Due to this, we show amazing dietary flexibility and adaptability due to our ability to eat a wide range of foodstuffs in most any environment we find ourselves in.
So “It is difficult to pinpoint what the human diet actually is … Nonetheless, humans are frequently described as omnivores” (Ulijaszek, Mann, and Elton, 2013: 59). Omnivores normally feed at two or more trophic levels, though others define it as just consuming plants and animals (Chubaty et al, 2014). Trophic level one is taken up by plants; level two is taken up by herbivores—primary consumers; level three is taken up by predators—who feed on the herbivores; level four or five is taken up by apex predators or carnivores; while the last level is also taken up by detrivores—those who feed on waste. Though, of course, “omnivory” is a continuum and not a category in and of itself. Humans eat primary producers (plants) and primary consumers (herbivores) and some secondary consumers (like fish), “although human omnivory may only be possible because of technological processing” (Ulijaszek, Mann, and Elton, 2013: 59). Other animals described as “omnivorous” eat only foods from one trophic level and only consume food from another level when needed.
Humans—as a species—rely on meat consumption. Fonseca-Azevedo and Herculano-Houzel (2012) showed that the energetic cost of a brain is directly related to the number of neurons in the brain. So, there were metabolic limitations in regard to brain and body size. The number of hours available to feed along with the low caloric yield of plant foods explains why great apes have such large bodies and small brains—which was probably overcome by erectus, who probably started cooking food around 1.5 mya. If we consumed only a diet of raw foods, then it would have taken us around 9 h/day to consume the calories we would need to power our brains—which is just not feasible. So it is unlikely that erectus—who was the first to have the human body plan and therefore the ability to run, which implies he would have needed higher quality energy—would have survived on a diet of raw plant foods since it would take so many hours to consume enough food to power their growing brains.
We can see that we are adapted to eating meat by looking at our intestines. Our small intestines are relatively long, whereas our long intestines are relatively short, which indicates that we became adapted to eating meat. Our “ability to eat significant quantities of meat and fish is a significant departure from the dietary norm of the haplorhine primates, especially for animals in the larger size classes.” Though “Humans share many features of their gut morphology with other primates, particularly the great apes, and have a gut structure that reflects their evolutionary heritage as plant, specifically ripe fruit, eaters” (Ulijaszek, Mann, and Elton, 2013: 63). Chimpanzees are not physiologically adapted to meat eating, which can be seen in the development of hypercholesterolemia along with vascular disease, even when controlled diets in captivity (Ford and Stanford, 2004).
When consuming a lot of protein, though, “rabbit starvation” needs to be kept in mind. Rabbit starvation is a type of malnutrition that arises from eating little to no fat and high amounts of protein. Since protein intake is physiologically demanding (it takes the most energy to process out of the three macros), Ben-Dor et al (2011) suggest a caloric ceiling of about 35 percent of kcal coming from protein. So erectus’ protein ceiling was 3.9 g/bw per day whereas for Homo sapiens it was 4.0 g/bw per day. Ben-Dor et al (2011) show that erectus’ DEE (daily energy expenditure) was about 2704 kcal, with “a maximum long-term plant protein ceiling of 1014 calories“, implying that erectus was, indeed, an omnivore. So, of course, the consumption of protein and raw plants are physiologically limited. Since erectus’ ceiling on protein intake was 947 kcal and his ceiling on raw plant intake was 1014 kcal, then, according to the model proposed by Ben-Dor et al (2011), erectus would have needed to consume about 744 kcal from fat, which is about 27 percent of his overall caloric intake and 44 percent of animal product intake.
Neanderthals would have consumed between 74-85 percent of their daily caloric energy during glacial winters from fat, with the rest coming from protein (Ben-Dor, Gopher, and Barkai, 2016), while consuming between 3,360 to 4,480 kcal per day (Steegman, Cerny, and Holliday, 2002). (See more on Neanderthal diet here.) Neanderthals consumed a large amount of protein, about 292 grams per day (Ben-Dor, Gopher, and Barkai, 2016: 370). Since our close evolutionary cousins (Neanderthals and erectus) ate large amounts of protein and fat, they were well-acclimated, physiologically speaking, to their high-protein diets. Though, their diets were not too high in protein to where rabbit starvation would occur—fat was consumed in sufficient amounts in the animals that Neanderthals hunted and killed, so rabbit starvation was not a problem for them. But since rabbit starvation is a huge problem for our species, “It is therefore unlikely that humans could be true carnivores in the way felids are” (Ulijaszek, Mann, and Elton, 2013: 66).
We consume a diet that is both omnivorous and eclectic, which is determined by our phylogeny through the form of our guts; we have nutritional diversity in our evolutionary history. We needed to colonize new lands and, since animals can only consume what is in their ecosystem, the foods that are edible in said ecosystem will be what is consumed by that animal. Being eclectic feeders made the migration out of Africa possible.
But humans are not true carnivores, contrary to some claims. “Meat-eating has allowed humans to colonize high latitudes and very open landscapes. However, bearing in mind the phylogenetic constraints that prevent humans from being true carnivores, such expansion was probably not accomplished through meat-eating alone. Instead, humans have used their ability to technologically harvest, produce, and consume a very wide range of foods to help exploit all major biomes” (Ulijaszek, Mann, and Elton, 2013: 67).
Humans, though, lack the gut specialization and dentition to process grasses efficiently. This means that our ancestors ate animals that ate these things, and therefore the C4 they consumed elevated the levels in the fossils we discovered. Information like this implies that our ancestors ate across a wide variety of trophic levels and had substantial dietary diversity throughout evolutionary history.
“Hominins lack the specialized dentition found in carnivorans (the group of animals that includes the cat and dog families) and other habitual meat and bone eaters, so must have pre-processed at least some of the meat in their diet” (Ulijaszek, Mann, and Elton, 2013: 81). This is where stone tools come into play (Zink and Lieberman, 2016). “Processing” food can be anything from taking out nutrients to changing how the food looks. We can look at “food processing” as a form of pre-digestion before consumption. The use of stone tools, and cooking, was imperative for us to begin the processing of meat and other foods. This gave us the ability to “pre-digest” our food before consumption, which increases the available energy in any food that is cooked/processed. For example, cooking denatures protein strands and breaks down the cell walls which gelatinizes the collagen in the meat which allows for easier chewing and digestion. Carmody et al (2016) showed that adaptation to a cooked diet began around 275 kya.
In his book Catching Fire, Wrangham (2009: 17-18) writes:
Raw-foodists are dedicated to eating 100 percent of their diets raw, or as close to 100 percent as they can manage. There are only three studies of their body weight, and all find that people who eat raw tend to be thin. The most extensive is the Giessen Raw Food study, conducted by nutritionist Corinna Koebnick and her colleagues in Germany, who used questionnaires to study 513 raw-foodists who ate from 70 to 100 percent of their diet raw. They chose to eat raw to be healthy, to prevent illness, to have a long life, or to live naturally. Raw food included not only uncooked vegetables and occasional meat, but also cold-pressed oil and honey, and some items were lightly heated such as dried fruits, dried meat, and dried fish. Body mass index (BMI), which measures weight in relation to the square of the height, was used as a measure of fatness. As the proportion of food eaten raw rose, BMI fell. The average weight loss when shifting from a cooked to a raw food diet was 26.5 pounds (12 kilograms) for women and 21.8 pounds (9.9 kilograms) for men. Among those eating a purely raw diet (31 percent), the body weights of almost a third indicated chronic energy deficiency. The scientists’ conclusion was unambiguous: “a strict raw food diet cannot guarantee an adequate energy supply.”
Also, vegetarians and meat-eaters who cooked their food have similar body weights. This implies that cooking food—no matter the type—gives more caloric energy to use for the body and that raw-foodists are fighting a losing battle with biology, consuming raw foods at such a high quantity that our guts are not used for. As can be seen above in the citation from Fonseca-Azevedo and Herculano-Houzel (2012), great apes who eat nothing but raw food have large guts and bodies which are needed to consume the raw plant foods they eat but we cannot thrive on such a diet because it is not calorically nor nutritionally viable for us—most importantly due to the size of our brains and its caloric requirements.
Carmody, Weintraub, and Wrangham (2011) show that modern raw-foodists who subsist on raw meat and plants have nutrient deficiencies and chronic energy deficiencies, even though they process their foods (cooking is a form of processing, as is cutting, mashing, pounding, etc) in different manners, while females experience low fecundity. Thus, the cooking of food seems to be needed for normal biological functioning; we have clearly evolved past consuming all raw foods. So it is clear that cooking—along with meat-eating—was imperative to our evolution. (Which does not mean that humans only ate meat and that eating meat and only meat is part of our evolutionary history.) Cooking food lead to it gelatinizing which denatured the protein, leading to easier mastication of the food, which meant less force since the food was not as hard after cooking. This then led to smaller teeth, over time, which was seen in erectus (Zink and Lieberman, 2016). This was due to cooking along with tool-use: the tool-use lead to smaller particles leading to less force per bite, which eventually led to smaller teeth in our lineage.
Finally, humans are said to be “facultative carnivores.” A facultative carnivore is an animal that does best on a carnivorous diet but can survive—not thrive—on other foodstuffs when meat is not available. This, though, doesn’t make sense. Humans are eclectic feeders—omnivorous in nature. Yes, we began cooking about 1.5 mya; yes meat-eating (and the cooking of said meat) is huge in the evolution of our species; yes without meat and cooking we would not have had the energy requirements to split off from chimpanzees/great apes. But this does not mean that we do “best” on a carnivorous diet. There are about 7,105 ethnic groups in the world (Spencer, 2014: 1029), and so to say that all of these ethnies would do the same or similar, physiologically speaking, on an all-meat diet is crazy talk. The claims that we subsisted on one type of food over the other throughout our evolutionary history is a bold claim—with no basis in evolutionary history.
Marlene Zuk (2013: 103-104), author of Paleofantasy writes:
Another implication of the importance Marlowe attaches to bow hunting is that, rather than starting out as exclusively carnivorous and then adding starches and other plant material to the diet, ancient humans have been able to increase the proportion of meat only after newer technology had come about, a mere 30,000 years ago. Other anthropologists concur that the amount of meat in the human diet grew as we diverged from our other primate ancestors. All of this means that, first, contrary to the claims of many paleo-diet proponents, the earliest humans did not have an exclusively meat-based diet that we are best adapted to eat; and second, our ancestors’ diets clearly changed dramatically and repeatedly over the last tens, not to mention hundreds, thousands of years, even before the advent of agriculture.
The assumption that we were fully (or even mostly) carnivorous and then added plant foods/carbs is clearly false. “Fantasies” like this are “just-so stories”; they are nice-sounding stories, but reality is clearly more nuanced than people’s evolutionary and Stone Age imaginations. This makes sense, though. Since we evolved from an LCA (last common ancestor) with chimpanzees some 6.3 mya (Patterson et al, 2006). So why would it make sense that we would then, ultimately, only subsist on an all-meat diet, if our LCA with chimpanzees was most likely a forager who lived in the trees (Lieberman, 2013).
One thing, though, I’m sure that everyone agrees with is that the environments we have constructed for ourselves in the first world are maladaptive—what is termed an “evolutionary mismatch” (Lieberman, 2013; Genne-Bacon, 2014). The mismatch arises from the high-carb food environments we have constructed, with cheap foodstuffs that is loaded with sugar, salt, and fat which is much more addictive than on their own (see Kessler, 2010). This makes food more palatable and people then want to eat it more. Foods like this, obviously, were not in our OEE (original evolutionary environment), and therefore cause us huge problems in our modern-day environments. Evolutionary mismatches occur when technological advancement increases faster than the genome can adapt. This can clearly be seen in our societies and the explosion of obesity over the past few decades (Fung, 2016, 2018).
We did not evolve eating highly processed carbohydrates loaded with salt and sugar. That much everyone can agree on.
Conclusion
It is clear that both claims from vegans/vegetarians and carnivores are false: there is no one “human diet” that we “should” be eating. Individual variation in different physiologic processes implies that there is no one “human diet”, no matter what type of food is being pushed as “what we should be” eating. Humans are eclectic feeders; we will eat anything since “Humans show remarkable dietary flexibility and adaptability“. Furthermore, we also “have a relatively unspecialized gut, with a colon that is shorter relative to overall size than in other apes; this is often attributed to the greater reliance on faunivory in humans (Chivers and Langer 1994)” (Ulijaszek, Mann, and Elton, 2013: 58). Our dietary eclectism can be traced back to our Australopithecine ancestors. The claim that we are either “vegetarian/vegan or carnivore” throughout our evolution is false.
Humans aren’t “natural carnivores” or “natural vegans/vegetarians.” Humans are eclectic feeders. Animals eat whatever is in their ecosystem. Ergo humans are omnivores, though we can’t pinpoint what the “human diet” is since there is great variability in it due to culture/ecology, we know one thing: we did not subsist on mainly only one food; we had a large variety of food, especially with fallback foods, to consume throughout our evolutionary history. So claims that we evolved to eat a certain way (as vegans/vegetarians and carnivores claim) is false. (Note I am not saying that high carb diets are good; I’ve railed hard on them.)
Nutrition, Development, Epigenetics, and Physical Plasticity
1650 words
Humans are extremely “plastic”. “Plastic” meaning that our development can be shaped by what goes on (or does not go in) in our developmental environment along with the environment outside of the womb. Many factors drive development, and if one factor changes then part of the developmental course for the organism changes as well. Thus, environment can and does drive development, with the addition (or subtraction) of different factors. In this article, I will discuss some of the factors that drive development and physical plasticity and what can change them.
Subsistence provides food while food provides nutrition. Nutrients, then, supply our bodies with energy and promote tissue growth—among other things. However, nutrient requirements vary across and between species, while all mammals need a mixture of macronutrients (carbs, fat, protein, water, and fiber) and micronutrients (vitamins and minerals). Biological variability in nutrient requirements and “the eventual degree of metabolic function that an individual can achieve for a particular intake level is determined to a greater or lesser extent by genetic variants in enzymes controlling the absorption, uptake, distribution, retention or utilization of the nutrient” (Molloy, 2004: 156). Thus, individuals who consume the same amount of micro and macronutrients—who also have different polymorphisms in genes coding for the metabolism of any nutrient (through hormones and enzymes)—can, and do, have differing physiological responses to same vitamin intake. Thus, differences in genetic polymorphisms between individuals can—and do—lead to different disease.
Next we have phenotypic plasticity. Phenotypic plasticity, simply put, is the ability for a genome to express a different phenotype in variable environments. For instance, people born in hotter environments—no matter their race or ethnicity—develop larger pores in order to sweat more, since sweating is needed for cooling the body (Lieberman, 2015). Phenotypic plasticity can be a problem, though, in environments with numerous environmental stressors that will stress the mother and, in turn, affect the baby’s development in the womb as well affecting post-birth events. An example of this is when food availability is low and exposure to infection is high (in-utero and post-birth), and when these stressors are removed, the organism in question shows “catch-up growth”, implying that these stressors impeded the development of the organism in question.
Maternal nutritional imbalance has been found—both in animal studies and epidemiological studies—and metabolic disturbances, during critical windows of development for the organism, have both a persistent effect on the health of the organism and can be transmitted epigenetically to future generations (Gallou-Kabani and Junien, 2005). Gallou-Kabani and Junien (2005) write:
Epigenetic chromatin marks may be propagated mitotically and, in some cases, meiotically, resulting in the stable inheritance of regulatory states. Transient nutritional stimuli occurring at critical ontogenic stages may have lasting influences on the expression of various genes by interacting with epigenetic mechanisms and altering chromatin conformation and transcription factor accessibility (11).
Thus, metabolic syndrome can show transgenerational effects by way of incomplete erasure of the epigenetic factors carried by grandparents and parents. (See also Treretola et al, 2005.) Epigenetic regulation was extremely important during our evolution and especially during the development of the human organism, and is how and why we are so phenotypically plastic.
Epigenetic regulation during fetal reprogramming of the individual in preparation for the environment they expect to enter is likely to be a response to seasonal energy imbalance; changes that favour the metabolic efficiency are likely to be adaptive in such circumstances. Removal of seasonal energy stress, as has taken place in contemporary industrialized societies, may turn efficiency toward pathology. Humans thus have evolved an animal model that can respond genetically (through natural selection), phenotypically (through developmental plasticity) and epigenetically (by a balance of both). (Ulijaszek, Mann, and Elton, 2013: 19)
This seems to be a fundamental response to the human organism in-utero, responding to the lack of food in its environment and growing accordingly (low birth weight, susceptibilities to differing disease), which are still a problem for much of the developed world. Though this can be maladaptive in the developed, industrialized world, since poor early-life environments can lead to epigenetic changes which then spell out bad consequences for the low-birth-weight baby who was exposed to a slew of negative nutritional factors during conception (and post-birth).
It has already been established that nutrition can alter the genome and epigenome (Niculescu and Lupu, 2011; Niculescu, 2012; Anderson, Sant, and Dolinoy, 2012). So if differing nutritional effects can alter the genome and epigenome and these effects are transgenerationally inherited by future generations, then famines change the expression of the genome and epigenome which can then inherited by future generations if the epigenetic factors carried by the grandparents and parents are not erased (and there is mounting evidence for this claim, see Yang, Liu, and Sun, 2017).
There is evidence of phenotypic plasticity regarding the lack of nutrition when it comes to humans, in-utero, and the evidence comes from the Dutch Family Studies (see Lumey et al, 2007 for an overview of the project). Individuals who were prenatally exposed to the Dutch winter famine of 1944-45, six decades later, had less DNA methylation of the IGF2 (insulin-like growth factor 2) gene than same-sex siblings who were not exposed to the winter famine (Heijmns et al, 2008). The IGF2 gene plays an essential role of the development of the fetus before birth. The gene is highly active during fetal development, but much less so after birth. (It should be noted that the loss of imprinting on the IGF2 gene can promote prostate cancer; Fenner, 2017 and loss of imprinting on IGF2 can also promote other types of cancer as well; Livingstone, 2013).
Stein et al (2009) concluded that “famine exposure prior to conception is associated with poorer self-reported mental health and a higher level of depressive symptoms.” Tobi et al (2009) write that their data “support the hypothesis that associations between early developmental conditions and health outcomes later in life may be mediated by changes in the epigenetic information layer.” Tobi et al (2014) also show that the “Epigenetic modulation of pathways by prenatal malnutrition may promote an adverse metabolic phenotype in later life.” The prenatal—and neonatal—periods of development are of utmost importance in order for the organism to develop normally, any deviation outside of these measures can—and does—affect the genome and epigenome (Hajj et al, 2014).
Another strong example that these responses are adaptive to the organism in question is the fact that people who were exposed to nutritional imbalances in the womb showed a higher chance of becoming obese later in life (Roseboom, de Rooji, and Painter, 2006). Their study has implications for babies born in developing countries (since famines mirror, in a way, developing countries). Roseboom, de Rooji, and Painter (2006: 489) write:
This may imply that adaptations that enable the fetus to continue to grow may nevertheless have adverse consequences for health in later life.
Roseboom, de Rooji, and Painter (2006: 490) also write:
The nutritional experience of babies who were exposed to famine in early gestation may resemble that of babies in developing countries whose mothers are undernourished in early pregnancy and receive supplementation later on, but also of babies in developed countries whose mothers suffer from severe morning sickness.
So on-going studies, such as the Dutch Famine Study, have the chance to elucidate the mechanisms of low birth weight, and it can also show us how and why those exposed to adverse conditions in the womb show so many negative symptoms which are not present in kin who were not exposed to such malnutrition in the womb. These findings also suggest that nutrition before—and after—pregnancy can play a role in disease acquisition later in life. The fact that those exposed to famines have a higher chance of becoming obese later in life (Abeleen et al, 2012; Meng et al, 2017) shows that this adaptive response of the organism in the womb was very important in our evolution; the babe exposed to low maternal nutrition in the womb can, after birth, consume enough energy to become overweight, which would have been an adaptive evolutionary response to low maternal caloric energy.
Babies who are exposed to maternal under-nutrition in the womb—when exposed to an environment with ample foodstuffs—are at heightened risk of becoming type II diabetics and acquiring metabolic syndromes (Robinson, Buchholz, and Mazurak, 2007). This seems to be an adaptive, plastic response of the organism: since nutrients/energy were in low quantity in the womb, low nutrients/energy in the womb changed the epigenome of the organism, and so when (if) the organism is exposed to an environment with ample amounts of food energy, they will then have a higher susceptibility to metabolic syndromes and weight gains, due to their uterine environment. (Diet also has an effect on brain plasticity in both animals and humans, in the womb and out of it; see Murphy, Dias, and Thuret, 2014.)
In sum, phenotypic plasticity, which is driven in part by epigenetics, was extremely important in our evolution. This epigenetic regulation that occurs in the womb prepared the individual in question to be able to respond to the energy imbalance of the environment the organism was born in. The plasticity of humans, and animals, in regard to what occurs (or does not occur) in the environment, is how we were able to survive in new environments (not ancestral to our species). Epigenetic changes that occur in the grandparental and parental generations, when not completely erased during the meiotic division of cells, can affect future generations of progeny in a negative way.
The implications of the data are clear: under-nutrition (and malnutrition) affect the genome and epigenome in ways that are inherited through the generations, which is due to the physical plasticity of the human in-utero as well as post-birth when the baby developing. These epigenetic changes then lead to the one who experienced the adverse uterine environment to have a higher chance of becoming obese later in life, which suggests that this is an adaptive response to low amounts of nutrients/caloric energy in the uterine environment.
Race and Vitamin D Deficiency
1600 words
Vitamin D is an important “vitamin” (it is really a steroid hormone). It is produced when the skin (the largest organ in the body) is exposed to the sun’s UVB rays (Nair and Maseeh, 2012). So this is one of the only ways to get natural levels of UVB. We can then think that, if a population is outside of its natural evolutionary habitat (the habitat where that skin color evolved), then we should note numerous problems caused by the lack of vitamin D in whichever population is studied outside of a location that doesn’t get the correct amount of UVB rays from the sun.
Black Americans are more likely than other ethnies to be deficient in vitamin D (Harris, 2006; Cosman et al, 2007; Nair, 2012; Forest and Stuhldreher, 2014; Taksler et al, 2014). But, paradoxically, low vitamin D levels don’t cause weaker bones in black Americans (O’Conner et al, 2014). However, like with all hypotheses, there are naysayers. For example. Powe et al (2013) argue that vitamin D tests misdiagnose blacks, that blacks have a form of the vitamin that cells can use called 25-hydroxyvitamin D. They conclude: “Community-dwelling black Americans, as compared with whites, had low levels of total 25-hydroxyvitamin D and vitamin D–binding protein, resulting in similar concentrations of estimated bioavailable 25-hydroxyvitamin D. Racial differences in the prevalence of common genetic polymorphisms provide a likely explanation for this observation.” Though there are a whole host of problems here.
The limitations of Powe et al (2013) striking: it was cross-sectional and observational (like most nutrition studies) so they were unable to predict effects of vitamin-D binding protein on bone fractures; no data on the consumption of vitamin D supplements; measurement of bone turnover markers, urinary calcium excretion and levels of 1,25-dihydroxyvitamin D may explain the effect of VDBP (vitamin D-binding protein) on mineral metabolism; and they relied on a calculation, rather than a measurement of 25-hydroxyvitamin D levels.
Powe et al’s (2013) findings, though, have been disputed. Using different measurement tools from Powe et al (2013), Henderson et al (2015) conclude that “Counter to prior observations by immunoassay, VDBG concentrations did not vary by race.” While Bouillon (2014) writes: In our view, black Americans, as compared with white Americans, have lower levels of not only total 25-hydroxyvitamin D but also free or bioavailable 25-hydroxyvitamin D.” And finally, Hollis and Bikle (2014) write: “Specifically, for any given physically measured level of bio-available 25-hydroxyvitamin D, the authors are overestimating bio-available 25-hydroxyvitamin D by 2 to 2.5 times owing to underestimation of vitamin D–binding protein in blacks.”
Either way, even if what Powe et al (2013) conclude is true, that would not mean that black Americans should not supplement with vitamin D, since many diseases and health problems are associated with low vitamin D intake in blacks, including osteoporosis, cardiovascular disease, cancer, diabetes, and other serious conditions (Harris, 2006). An indirect relationship between low levels of vitamin D and hypertension is also noted (Mehta and Agarwal, 2017). Since there is an indirect relationship between vitamin D levels and hypertension, then we should keep an eye on this because black Americans have some of the highest levels of hypertension in the world (Ferdinand and Armani, 2007; see also Fuchs, 2011).
Vitamin D is, of course, important for skeletal and nonskeletal health (Kennel et al, 2010). So if vitamin D is important for skeletal and nonskeletal health, we should see more diseases in black Americans that imply a lack of this steroid in the body. Although blacks have stronger bones even when deficient in vitamin D, it is still observed that black children who break their forearms have less vitamin D circulating in their blood (Ryan et al, 2011). This observation is borne out by the data, since black children are more likely to be deficient in vitamin D compared to other ethnies (Moore, Murphy, and Hollick, 2005). Since black skin predicts vitamin D deficiency (Thomas and Demay, 2000), it seems logical to give vitamin D supplements to children, especially black children, on the basis that it would help lower incidences of bone fractures, even though blacks have stronger bones than whites.
Furthermore, physiologically “normal” levels of vitamin D differ in blacks compared to whites (Wright et al, 2012). They showed that it is indeed a strong possibility that both whites and blacks have different levels of optimum vitamin D. Wright et al (2012) showed that there is a relationship between 25(OH)D levels and intact parathyroid hormone (iPth); for blacks, the threshold in which there was no change was 20 ng/ml whereas for whites it was 30 ng/ml which suggests that there are different levels of optimal vitamin D for each race, and the cause is due to skin color. Thus, physiologically “normal” levels of vitamin D differ for blacks and whites.
There is also a high prevalence of vitamin D deficiency/insufficiency and asthma in black inner-city youth in Washington DC (Freishtat et al, 2010). We can clearly see that, even though black Americans have stronger bones than white Americans and vitamin D predicts bone strength, the fact that blacks have stronger bones than whites even while being deficient in vitamin D on average does not mean that black Americans should not supplement with vitamin D, since it would ameliorate many other problems they have that are related to vitamin D deficiency.
There are also racial differences in prostate cancer (PCa) acquisition too, and vitamin D deficiency may also explain this disparity (Khan and Partin, 2004; Bhardwaj et al, 2017). I have heavily criticized the explanations that testosterone influences PCa, while having indicated that environmental factors such as diet and vitamin D deficiency may explain a large amount of the gap (Batai et al, 2017; but see Stranaland et al, 2017 for a contrary view). Since low vitamin D is related to prostate cancer, by supplementing with vitamin D, it is possible that levels of PCa may decrease. Kristal et al (2014) show that both high and low levels of vitamin D are associated with PCa.
Evidence also exists that vitamin D levels and hypertension are related. Rostand (2010) proposes a unified hypothesis: an important role exists in vitamin D deficiency and the pathogenesis and maintenance of hypertension in blacks (Rostand, 2010).
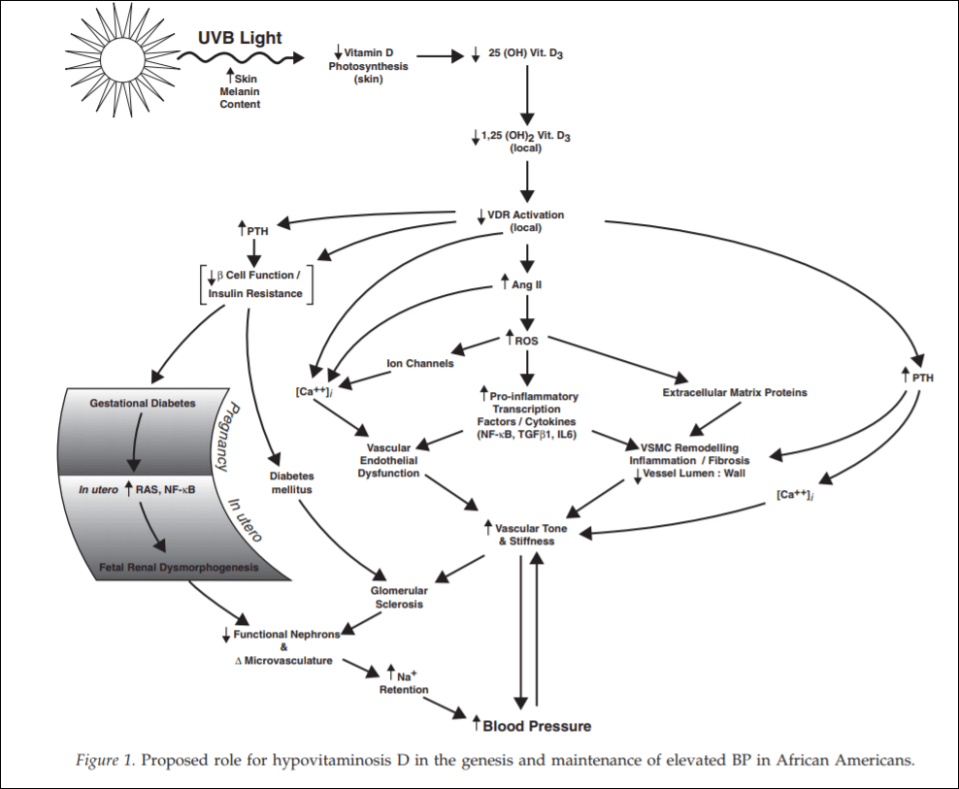
(From Rostand, 2010)
Since black Americans are no longer near the equator, their ability to synthesize vitamin D from UVB rays is diminished. This then probably leads the RAS (renin-angiotensin system) and inflammatory cytokine activation which then leads to vascular endothelial dysfunction along with structural changes to the microvasculature, which have been linked to vascular (arterial) stiffness along with increased vascular resistance, and these changes are shown to precede hypertension, which also occurs early in life. So since blacks are deficient in vitamin D, which even starts in the womb (Bodnar et al, 2007; Dawodu and Wagner, 2007; Lee et al, 2007; Khalessi et al, 2015; Seto et al, 2016), and this vitamin D deficiency most likely produces changes in large and small arteries and arterials, this could be the explanation for higher hypertension in black Americans (Rostand, 2010: 1701).
This would be a large environmental mismatch: since the population is displaced from its ancestral homeland, then this causes problems since it is not the environment where their ancestors evolved. So in this case, since black Americans are concentrated in the southeast corner of the United States, this may explain the high rates of vitamin D deficiency and hypertension in the black American community.
People whose ancestors evolved in locations with fewer UVB rays have lighter skin, whereas people whose ancestors evolved in locations with more UVB rays have darker skin. Thus, by placing populations in their opposite evolutionary environment, we can see how and why deleterious effects would occur in the population that is in the mismatched environment. For whites, skin cancer would occur, whereas for blacks, higher rates of hypertension and low birth weights occur.
Looking at levels of vitamin D deficiency in races is a great way to understand the evolution of certain populations. Because if the vitamin D hypothesis is correct, if skin color is an adaptation to UVB rays, with light skin being an adaptation to low UVB while dark skin is an adaptation to high UVB, then we can safely hypothesize about certain problems that would arise in races that are outside of their natural habitats. We have confirmed these hypotheses—black Americans who are outside of the location that their ancestors evolved in are more likely to have deleterious symptoms, and the symptoms are due to differences in vitamin D production, which come down to differences in skin color and how the skin synthesizes vitamin D in low-light environments.
Even though blacks have stronger bones than whites, this does not mean that they do not experience fractures at a high rate—especially children—and since the association was noticed, then by supplementing with vitamin D, this may lower the disparity of these types of injuries.
Since black Americans, compared to their evolutionary history, live in low-light environments, this then explains the how and why of vitamin D deficiency and why blacks need to supplement with vitamin D; no matter if certain studies show that blacks are ‘healthy’ even though they have low levels of vitamin D. If true (which I strongly doubt), that does not mean that black Americans should not supplement with vitamin D, because numerous other maladies are associated with vitamin D intake. This is one aspect where understanding the evolution of our species and the different races in it would lead to better medical care for individuals and ancestral groups that may need special treatment.
It is clear that race and geography should inform vitamin D intake, for if we do this, many diseases that arise can be ameliorated and quality of life can increase for everyone.
Nutrition and Antisocial Behavior
2150 words
What is the relationship between nutrition and antisocial behavior? Does not consuming adequate amounts of vitamins and minerals lead to an increased risk for antisocial behavior? If it does, then lower class people will have commit crimes at a higher rate, and part of the problem may indeed be dietary. Though, what kind of data is there that lends credence to the idea? It is well-known that malnutrition leads to antisocial behavior, but what kind of effect does it have on the populace as a whole?
About 85 percent of Americans lack essential vitamins and minerals. Though, when most people think of the word ‘malnutrition’ and the imagery it brings along with it, they assume that someone in a third-world country is being talked about, say a rail-thin kid somewhere in Africa who is extremely malnourished due to lack of kcal and vitamins and minerals. However, just because one lives in a first-world country and has access to kcal to where they’re “not hungry” doesn’t mean that vitamin and mineral deficiencies do not exist in these countries. This is known as “hidden hunger” when people can get enough kcal for their daily energy needs but what they are eating is lower-quality food, and thus, they become vitamin and nutrient deficient. What kind of effects does this have?
Infants are most at risk, more than half of American babies are at-risk for malnutrition; malnutrition in the postnatal years can lead to antisocial behavior and a lower ‘IQ’ (Galler and Ramsey, 1989; Liu et al, 2003; Galler et al, 2011, 2012a, 2012b; Gesch, 2013; Kuratko et al, 2013; Raine et al, 2015; Thompson et al, 2017). Clearly, not getting pertinent vitamins and minerals at critical times of development for infants leads to antisocial behavior in the future. These cases, though, can be prevented with a good diet. But the preventative measures that can prevent some of this behavior has been demonized for the past 50 or so years.
Poor nutrition leads to the development of childhood behavior problems. As seen in rat studies, for example, lack of dietary protein leads to aggressive behavior while rats who are protein-deficient in the womb show altered locomotor activity. The same is also seen with vitamins and minerals; monkeys and rats who were fed a diet low in tryptophan were reported to be more aggressive whereas those that were fed high amounts of tryptophan were calmer. Since tryptophan is one of the building blocks of serotonin and serotonin regulates mood, we can logically state that diets low in tryptophan may lead to higher levels of aggressive behavior. The role of omega 3 fatty acids are mixed, with omega 3 supplementation showing a difference for girls, but not boys (see Itomura et al, 2005). So, animal and human correlational studies and human intervention studies lend credence to the hypothesis that malnutrition in the womb and after birth leads to antisocial behavior (Liu and Raine, 2004).
We also have data from one randomized, placebo-controlled trial showing the effect of diet and nutrition on antisocial behavior (Gesch et al, 2002). They state that since there is evidence that offenders’ diets are lacking in pertinent vitamins and minerals, they should test whether or not the introduction of physiologically adequate vitamins, minerals and essential fatty acids (EFAs) would have an effect on the behavior of the inmates. They undertook an experimental, double-blind, placebo-controlled randomized trial on 231 adult prisoners and then compared their write-ups before and after nutritional intervention. The vitamin/mineral supplement contained 44 mg of DHA (omega 3 fatty acid docosahexaenoic acid; plays a key role in enhancing brain structure and function, stimulating neurite outgrowth), 80 mg of EPA (eicosapentaenoic acid; n3), and 1.26 g of ALA (alpha-linolenic acid), 1260mg of LA (linolic acid), and 160mg of GLA (gamma-Linolenic acid, n6) and a vegetable oil placebo. (Also see Hibbeln and Gow, 2015 for more information on n3 and nutrient deficits in childhood behavior disorders and neurodevelopment.)
Raine (2014: 218-219) writes:
We can also link micronutrients to specific brain structures involved in violence. The amygdala and hippocampus, which are impaired in offenders, are packed with zinc-containing neurons. Zinc deficiency in humans during pregnancy can in turn impair DNA, RNA, and protein synthesis during brain development—the building blocks of brain chemistry—and may result in very early brain abnormalities. Zinc also plays a role in building up fatty acids, which, as we have seen, are crucial for brain structure and function.
Gesch et al (2002) found pretty interesting results: those who were given the capsules with vitamins, minerals, and EFAs had 26.3 percent fewer offenses than those who got the placebo. Further, when compared with the baseline, when taking the supplement for two weeks, there was an average 35.1 percent reduction in offenses compared to the placebo group who showed little change. Gesch et al (2002) conclude:
Antisocial behaviour in prisons, including violence, are reduced by prisons, are reduced by vitamins, minerals and essential fatty acids with similar implications for those eating poor diets in the community.
Of course one could argue that these results would not transfer over to the general population, but to a critique like this, the observed effect of behavior is physiological; so by supplementing the prisoners’ diets giving them pertinent vitamins, minerals and EFAs, violence and antisocial behavior decreased, which shows some level of causation between nutrition/nutrient/fatty acid deprivation and antisocial behavior and violent activity.
Gesch et al (2002) found that some prisoners did not know how to construct a healthy diet nor did they know what vitamins were. So, naturally, since some prisoners didn’t know how to construct diets with an adequate amount of EFAs, vitamins and minerals, they were malnourished, though they consumed an adequate amount of calories. The intervention showed that EFA, vitamin and mineral deficiency has a causal effect on decreasing antisocial and violent behavior in those deficient. So giving them physiological doses lowered antisocial behavior, and since it was an RCT, social and ethnic factors on behavior were avoided.
Of course (and this shouldn’t need to be said), I am not making the claim that differences in nutrition explain all variance in antisocial and violent behavior. The fact of the matter is, this is causal evidence that lack of vitamin, mineral and EFA consumption has some causal effect on antisocial behavior and violent tendencies.
Schoenthaler et al (1996) also showed how correcting low values of vitamins and minerals in those deficient led to a reduction in violence among juvenile delinquents. Though it has a small n, the results are promising. (Also see Zaalberg et al, 2010.) These simple studies show how easy it is to lower antisocial and violent behavior: those deficient in nutrients just need to take some vitamins and eat higher-quality food and there should be a reduction in antisocial and violent behavior.
Liu, Zhao, and Reyes (2015) propose “a conceptual framework whereby epigenetic modifications (e.g., DNA methylation) mediate the link between micro- and macro-nutrient deficiency early in life and brain dysfunction (e.g., structural aberration, neurotransmitter perturbation), which has been linked to development of behavior problems later on in life.” Their model is as follows: macro- and micro-nutrient deficiencies are risk-factors for psychopathologies since they can lead to changes in the epigenetic regulation of the genome (along with other environmental variables such as lead consumption, which causes abnormal behavior and also epigenetic changes which can be passed through the generations; Senut et al, 2012; Sen et al, 2015) which then leads to impaired brain development, which then leads to externalizing behavior, internalizing behavior and autism and schizophrenia (two disorders which are also affected by the microbiome; Strati et al, 2017; Dickerson, 2017).
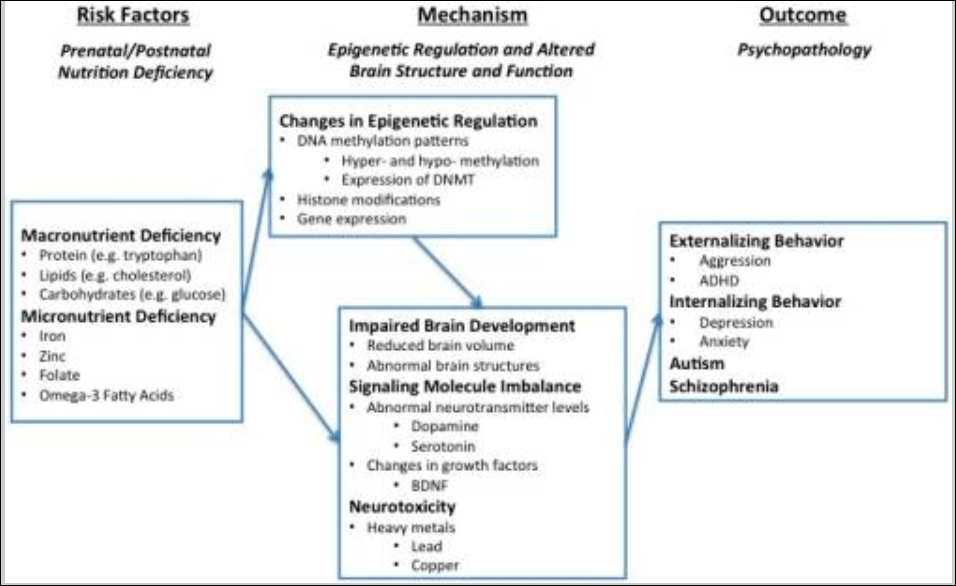
Clearly, since the food we eat gives us access to certain fatty acids that cannot be produced de novo in the brain or body, good nutrition is needed for a developing brain and if certain pertinent vitamins, minerals or fatty acids are missing, negative outcomes could occur for said individual in the future due to lack of brain development from being nutrient, vitamin, and mineral deficient in childhood. Further, interactions between nutrient deficiencies and exposure to toxic chemicals may be a cause of a large amount of antisocial behavior (Walsh et al, 1997; Hubbs-Tait et al, 2005; Firth et al, 2017).
Looking for a cause for this interaction between metal consumption and nutrient deficiencies, Liu, Zhao, and Reyes (2015) state that since protein and fatty acids are essential to brain growth, lack of consumption of pertinent micro- and macro-nutrients along with consumption of high amounts of protein both in and out of the womb contribute to lack of brain growth and, at adulthood, explains part of the difference in antisocial behavior. What you can further see from the above studies is that metals consumed by an individual can interact with the nutrient deficiencies in said individual and cause more deleterious outcomes, since, for example, lead is a nutrient antagonist—that is, it inhibits the physiologic actions of whatever bioavailable nutrients are available to the body for us.
Good nutrition is, of course, imperative since it gives our bodies what it needs to grow and develop as we grow in the womb, as adolescents and even into old age. So, therefore, developing people who are nutrient deficient will have worse behavioral outcomes. Further, lower class people are more likely to be nutrient deficient and consume lower quality diets than higher, more affluent classes, though it’s hard to discover which way the causation goes (Darmon and Drewnowski, 2008). Of course, the logical conclusion is that being deficient in vitamins, minerals and EFAs causes changes to the epigenome and retards brain development, therefore this has a partly causal effect on future antisocial, violent and criminal behavior. So, some of the crime difference between classes can be attributed to differences in nutrition/toxic metal exposure that induces epigenetic changes that change the structure of the brain and doesn’t allow full brain development due to lack of vitamins, minerals, and EFAs.
There seems to be a causal effect on criminal, violent and antisocial behavior regarding nutrient deficiencies in both juveniles and adults (which starts in the womb and continues into adolescence and adulthood). However, it has been shown in a few randomized controlled trials that nutritional interventions decrease some antisocial behavior, with the effect being strongest for those individuals who showed worse nutrient deficiencies.
If the relationship between nutrition/interaction between nutrient deficiencies and toxins can be replicated successfully then this leads us to one major question: Are we, as a society, in part, causing some of the differences in crime due to how our society is regarding nutrition and the types of food that are advertised to our youth? Are people’s diets which lead to nutrient deficiencies a driving factor in causing crime? The evidence so far on nutrition and its effects on the epigenome and its effects on the growth of the brain in the womb and adolescence requires us to take a serious look at this relationship. That lower class people are exposed to more neurotoxins such as lead (Bellinger, 2008) and are more likely to be nutrient deficient (Darmon and Drewnowski, 2008; Hackman, Farrah, and Meaney, 2011) then if they were educated on which foods to eat to avoid nutrient deficiencies along with avoiding neurotoxins such as lead (which exacerbate nutrient deficiencies and cause crime), then a reduction in crime should occur.
Nutrition is important for all living beings; and as can be seen, those who are deficient in certain nutrients and have less access to good, whole, nutritious food (who also have an increased risk for exposure to neurotoxins) can lead to negative outcomes. These things can be prevented, it seems, with a few vitamins/minerals/EFA consumption. The effects of sleep, poor diet (which also lead to metabolic syndromes) can also exacerbate this relationship, between individuals and ethnicities. The relationship between violence and antisocial behavior and nutrient deficiencies/the interaction with nutrient deficiencies and neurotoxins is a great avenue for future research to reduce violent crime in our society. Lower class people, of course, should be the targets of such interventions since there seems to be a causal effect—-however small or large—on behavior, both violent and nonviolent—and so nutrition interventions should close some of the crime gaps between classes.
Conclusion
The logic is very simple: nutrition affects mood (Rao et al, 2008; Jacka, 2017) which is, in part, driven by the microbiome’s intimate relationship with the brain (Clapp et al, 2017; Singh et al, 2017); nutrition also affects the epigenome and the growth and structure of the brain if vitamin and mineral needs are not met by the growing body. This then leads to differences in gene expression due to the foods consumed, the microbiome (which also influences the epigenome) further leads to differences in gene expression and behavior since the two are intimately linked as well. Thus, the aetiology of certain behaviors may come down to nutrient deficiencies and complex interactions between the environment, neurotoxins, nutrient deficiencies and genetic factors. Clearly, we can prevent this with preventative nutritional education, and since lower class people are more likely to suffer the most from these problems, the measures targeted to them, if followed through, will lower incidences of crime and antisocial/violent behavior.
Calories are not Calories
1300 words
More bullocks from Dr. Thompson:
I say that if you are over-weight and wish to lose weight, then you should eat less. You should keep eating less until you achieve your desired weight, and then stick to that level of calorific intake.
Why only talk about calories and assume that they do the same things once ingested into the body? See Feinman and Fine (2004) to see how and why that is fallacious. This was actually studied. Contestants on the show The Biggest Loser were followed after they lost a considerable amount of weight. They followed the same old mantra: eat less, and move more. Because if you decrease what is coming in, and expend more energy then you will lose weight. Thermodynamics, energy in and out, right? That should put one into a negative energy balance and they should lose weight if they persist with the diet. And they did. However, what is going on with the metabolism of the people who lost all of this weight, and is this effect more noticeable for people who lost more weight in comparison to others?
Fothergill et al (2016) found that persistent metabolic slowdown occurred after weight loss, the average being a 600 kcal slowdown. This is what the conventional dieting advice gets you, a slowed metabolism with you having to eat fewer kcal than one who was never obese. This is what the ‘eat less, move more’ advice, the ‘CI/CO’ advice is horribly flawed and does not work!
He seems to understand that exercise does not work to induce weight loss, but it’s this supposed combo that’s supposed to be effective, a kind of one-two punch, and you only need to eat less and move more if you want to lose weight! This is horribly flawed. He then shows a few table from a paper he authored with another researcher back in 1974 (Bhanji and Thompson, 1974).
Say you take 30 people who weigh the same, have the same amount of body fat and are the same height, they eat the same exact macronutrient composition, with the same exact foods, eating at a surplus deficit with the same caloric content, and, at the end of say, 3 months, you will get a different array of weight gained/stalled/decrease in weight. Wow. Something like this would certainly disprove the CI/CO myth. Aamodt (2016: 138-139) describes a study by Bouchard and Tremblay (1997; warning: twin study), writing:
When identical twins, men in their early 20s, were fed a thousand extra calories per day for about three months, each pair showed similar weight gains. In contrast, the gains varied across twin pairs, ranging from nine to twenty-nine pound, even though the calorie imbalance esd the same for everyone. An individual’s genes also influence weight loss. When another group of identical twins burned a thousand more calories per day through exercise while maintaining a stable food intake in an inpatient facility, their losses ranged from two to eighteen pounds and were even more similar within twin pairs than weight gain.
Take a moment to think about that. Some people’s bodies resis weight loss so well that burning an extra thousand calpires a day for three months, without eating more, leads them to lose only two pounds. The “weight loss is just math” crows we met in the last chapter needs to look at what happens when their math is applied to living people. (We know what usually happens: they accuse the poor dieter of cheating, whether or not it’s true.) If cutting 3,500 calories equals one pound of weight loss, then everyone on the twuns’ exercist protocol should have lost twenty-four pounds, but not a single participant lost that much. The average weight loss was only eleven pounds, and the individual variation was huge. Such differences can result from genetic influences on resting metabolism, which varies 10 to 15 percent between people, or from differences in the gut. Because the thousand-calorie energy imbalance was the same in both the gain and loss experiments, this twin research also illustrates that it’s easier to gain weight than to lose it.
That’s weird. If a calorie were truly a calorie, then, at least in the was CI/COers word things, everyone should have had the same or similar weight loss, not with the average weight loss less than half what should have been expected from the kcal they consumed. That is a shot against the CI/CO theory. Yet more evidence against comes from the Vermont Prison Experiment (see Salans et al, 1971). In this experiment, they were given up to 10,000 kcal per day and they, like in the other study described previously, all gained differing amounts of weight. Wow, almost as if individuals are different and the simplistic caloric math of the CI/COers doesn’t size up against real-life situations.
The First Law of Thermodynamics always holds, it’s just irrelevant to human physiology. (Watch Gary Taubes take down this mythconception too; not a typo.) Think about an individual who decreases total caloric intake from 1500 kcal per day to 1200 kcal per day over a certain period of time. The body is then forced to drop its metabolism to match the caloric intake, so the metabolic system of the human body knows when to decrease when it senses it’s getting less intake, and for this reason the First Law is not violated here, it’s irrelevant. The same thing also occurred to the Biggest Loser contestants. Because the followed the CI/CO paradigm of ‘eat less and move more’.
Processed food is not bad in itself, but it is hard to monitor what is in it, and it is probably best avoided if you wish to lose weight, that is, it should not be a large part of your habitual intake.
If you’re trying to lose weight you should most definitely avoid processed foods and carbohydrates.
In general, all foods are good for you, in moderation. There are circumstances when you may have to eat what is available, even if it is not the best basis for a permanent sustained diet.
I only contest the ‘all foods are good for you’ part. Moderation, yes. But in our hedonistic world we live in today with a constant bombardment of advertisements there is no such thing as ‘moderation’. Finally, again, willpower is irrelevant to obesity.
I’d like to know the individual weight gains in Thompson’s study. I bet it’d follow both what occurred in the study described by Aamodt and the study by Sims et al. The point is, human physiological systems are more complicated than to attempt to break down weight loss to only the number of calories you eat, when not thinking of what and how you eat it. What is lost in all of this is WHEN is a good time to eat? People continuously speak about what to eat, where to eat, how to eat, who to eat with but no one ever seriously discusses WHEN to eat. What I mean by this is that people are constantly stuffing their faces all day, constantly spiking their insulin which then causes obesity.
The fatal blow for the CI/CO theory is that people do not gain or lose weight at the same rate (I’d add matched for height, overall weight, muscle mass and body fat, too) as seen above in the papers cited. Why people still think that the human body and its physiology is so simple is beyond me.
Hedonism along with an overconsumption of calories consumed (from processed carbohydrates) is why we’re so fat right now in the third world and the only way to reverse the trend is to tell the truth about human weight loss and how and why we get fat. CI/CO clearly does not work and is based on false premises, no matter how much people attempt to save it. It’s highly flawed and assumed that the human body is so ‘simple’ as to not ‘care’ about the quality of the macro nor where it came from.
Race, Testosterone, Aggression, and Prostate Cancer
4050 words
Race, aggression, and prostate cancer are all linked, with some believing that race is the cause of higher testosterone which then causes aggression and higher rates of crime along with maladies such as prostate cancer. These claims have long been put to bed, with a wide range of large analyses.
The testosterone debate regarding prostate cancer has been raging for decades and we have made good strides in understanding the etiology of prostate cancer and how it manifests. The same holds true for aggression. But does testosterone hold the key to understanding aggression, prostate cancer and does race dictate group levels of the hormone which then would explain some of the disparities between groups and individuals of certain groups?
Prostate cancer
For decades it was believed that heightened levels of testosterone caused prostate cancer. Most of the theories to this day still hold that large amounts of androgens, like testosterone and it’s metabolic byproduct dihydrotestosterone, are the two many factors that drive the proliferation of cells and therefore, if a male is exposed to higher levels of testosterone throughout their lives then they are at a high risk of prostate cancer compared to a man with low testosterone levels, so the story goes.
In 1986 Ronald Ross set out to test a hypothesis: that black males were exposed to more testosterone in the womb and this then drove their higher rates of prostate cancer later in life. He reportedly discovered that blacks, after controlling for confounds, had 15 percent higher testosterone than whites which may be the cause of differential prostate cancer mortality between the two races (Ross et al, 1986) This is told in a 1997 editorial by Hugh McIntosh. First, the fact that black males were supposedly exposed to more testosterone in the womb is brought up. I am aware of one paper discussing higher levels of testosterone in black women compared to white women (Perry et al, 1996). Though, I’ve shown that black women don’t have high levels of testosterone, not higher than white women, anyway (see Mazur, 2016 for discussion). (Yes I changed my view on black women and testosterone, stop saying that they have high levels of testosterone it’s just not true. I see people still link to that article despite the long disclaimer at the top.)
Alvarado (2013) discusses Ross et al (1986), Ellis and Nyborg (1992) (which I also discussed here along with Ross et al) and other papers discussing the supposed higher testosterone of blacks when compared to whites and attempts to use a life history framework to explain higher incidences of prostate cancer in black males. He first notes that nutritional status influences testosterone production which should be no surprise to anyone. He brings up some points I agree with and some I do not. For instance, he states that differences in nutrition could explain differences in testosterone between Western and non-Western people (I agree), but that this has no effect within Western countries (which is incorrect as I’ll get to later).
He also states that ancestry isn’t related to prostate cancer, writing “In summation, ancestry does not adequately explain variation among ethnic groups with higher or lower testosterone levels, nor does it appear to explain variation among ethnic groups with high or low prostate cancer rates. This calls into question the efficacy of a disease model that is unable to predict either deleterious or protective effects.”
He then states that SES is negatively correlated with prostate cancer rates, and that numerous papers show that people with low SES have higher rates of prostate cancer mortality which makes sense, since people in a lower economic class would have less access to and a chance to get good medical care to identify problems such as prostate cancer, including prostate biopsies and checkups to identify the condition.
He finally discusses the challenge hypothesis and prostate cancer risk. He cites studies by Mazur and Booth (who I’ve cited in the past in numerous articles) as evidence that, as most know, black-majority areas have more crime which would then cause higher levels of testosterone production. He cites Mazur’s old papers showing that low-class men, no matter if they’re white or black, had heightened levels of testosterone and that college-educated men did not, which implies that the social environment can and does elevate testosterone levels and can keep them heightened. Alvarado concludes this section writing: “Among Westernized men who have energetic resources to support the metabolic costs associated with elevated testosterone, there is evidence that being exposed to a higher frequency of aggressive challenges can result in chronically elevated testosterone levels. If living in an aggressive social environment contributes to prostate cancer disparities, this has important implications for prevention and risk stratification.” He’s not really wrong but on what he is wrong I will discuss later on this section. It’s false that testosterone causes prostate cancer so some of this thesis is incorrect.
I rebutted Ross et al (1986) December of last year. The study was hugely flawed and, yet, still gets cited to this day including by Alvarado (2013) as the main point of his thesis. However, perhaps most importantly, the assay times were done ‘when it was convenient’ for the students which were between 10 am and 3 pm. To not get any wacky readings one most assay the individuals as close to 8:30 am as possible. Furthermore, they did not control for waist circumference which is another huge confound. Lastly, the sample was extremely small (50 blacks and 50 whites) and done on a nonrepresentative sample (college students). I don’t think anyone can honestly cite this paper as any evidence for blacks having higher levels of testosterone or testosterone causing prostate cancer because it just doesn’t do that. (Read Race, Testosterone and Prostate Cancer for more information.)
What may explain prostate cancer rates if not for differences in testosterone like has been hypothesized for decades? Well, as I have argued, diet explains a lot of the variation between races. The etiology of prostate cancer is not known (ACA, 2016) but we know that it’s not testosterone and that diet plays a large role in its acquisition. Due to their dark skin, they need more sunlight than do whites to synthesize the same amount of vitamin D, and low levels of vitamin D in blacks are strongly related to prostate cancer (Harris, 2006). Murphy et al (2014) even showed, through biopsies, that black American men had higher rates of prostate cancer if they had lower levels of vitamin D. Lower concentrations of vitamin D in blacks compared to whites due to dark pigmentation which causes reduced vitamin D photoproduction and may also account for “much of the unexplained survival disparity after consideration of such factors as SES, state at diagnosis and treatment” (Grant and Peiris, 2012).
Testosterone
As mentioned above, testosterone is assumed to be higher in certain races compared to others (based on flawed studies) which then supposedly exacerbates prostate cancer. However, as can be seen above, a lot of assumptions go into the testosterone-prostate cancer hypothesis which is just false. So if the assumptions are false about testosterone, mainly regarding racial differences in the hormone and then what the hormone actually does, then most of their claims can be disregarded.
Perhaps the biggest problem is that Ross et al is a 32-year-old paper (which still gets cited favorably despite its huge flaws) while our understanding of the hormone and its physiology has made considerable progress in that time frame. So it’s in fact not so weird to see papers like this that say “Prostate cancer appears to be unrelated related to endogenous testosterone levels” (Boyle et al, 2016). Other papers also show the same thing, that testosterone is not related to prostate cancer (Stattin et al, 2004; Michaud, Billups, and Partin, 2015). This kills a lot of theories and hypotheses, especially regarding racial differences in prostate cancer acquisition and mortality. So, what this shows is that even if blacks did have 15 percent higher serum testosterone than whites as Ross et al, Rushton, Lynn, Templer, et al believed then it wouldn’t cause higher levels of prostate cancer (nor aggression, which I’ll get into later).
How high is testosterone in black males compared to white males? People may attempt to cite papers like the 32-year-old paper by Ross et al, though as I’ve discussed numerous times the paper is highly flawed and should therefore not be cited. Either way, levels are not as high as people believe and meta-analyses and actual nationally representative samples (not convenience college samples) show low to no difference, and even the low difference wouldn’t explain any health disparities.
One of the best papers on this matter of racial differences in testosterone is Richard et al (2014). They meta-analyzed 15 studies and concluded that the “racial differences [range] from 2.5 to 4.9 percent” but “this modest difference is unlikely to explain racial differences in disease risk.” This shows that testosterone isn’t as high in blacks as is popularly misconceived, and that, as I will show below, it wouldn’t even cause higher rates of aggression and therefore criminal behavior. (Rohrmann et al 2007 show no difference in testosterone between black and white males in a nationally representative sample after controlling for lifestyle and anthropometric variables. Whereas Mazur, 2009 shows that blacks have higher levels of testosterone due to low marriage rates and lower levels of adiposity, while be found a .39 ng/ml difference between blacks and whites aged 20 to 60. Is this supposed to explain crime, aggression, and prostate cancer?)
However, as I’ve noted last year (and as Alvarado, 2013 did as well), young black males with low education have higher levels of testosterone which is not noticed in black males of the same age group but with more education (Mazur, 2016). Since blacks of a similar age group have lower levels of testosterone but are more highly educated then this is a clue that education drives aggression/testosterone/violent behavior and not that testosterone drives it.
Mazur (2016) also replicated Assari, Caldwell, and Zimmerman’s (2014) finding that “Our model in the male sample suggests that males with higher levels of education has lower aggressive behaviors. Among males, testosterone was not associated with aggressive behaviors.” I know this is hard for many to swallow that testosterone doesn’t lead to aggressive behavior in men, but I’ll cover that in the last and final section.
So it’s clear that the myth that Rushton, Lynn, Templer, Kanazawa, et al pushed regarding hormonal differences between the races are false. It’s also with noting, as I did in my response to Rushton on r/K selection theory, that the r/K model is literally predicated on 1) testosterone differences between races being real and in the direction that Rushton and Lynn want because they cite the highly flawed Ross et al (1986) and 2) testosterone does not cause higher levels of aggression (which I’ll show below) which then lead to higher rates of crime along with higher rates of incarceration.
A blogger who goes by the name of ethnicmuse did an analysis of numerous testosterone papers and he found:
 Which, of course, goes against a ton of HBD theory, that is, if testosterone did what HBDers believed it does (it doesn’t). This is what it comes down to: blacks don’t have higher levels of testosterone than whites and testosterone doesn’t cause aggression nor prostate cancer so even if this relationship was in the direction that Rushton et al assert then it still wouldn’t cause any of the explanatory variables they discuss.
Which, of course, goes against a ton of HBD theory, that is, if testosterone did what HBDers believed it does (it doesn’t). This is what it comes down to: blacks don’t have higher levels of testosterone than whites and testosterone doesn’t cause aggression nor prostate cancer so even if this relationship was in the direction that Rushton et al assert then it still wouldn’t cause any of the explanatory variables they discuss.
Last year Lee Ellis published a paper outlining his ENA theory (Ellis, 2017). I responded to the paper and pointed out what he got right and wrong. He discussed strength (blacks aren’t stronger than whites due to body type and physiology, but excel in other areas); circulating testosterone, umbilical cord testosterone exposure; bone density and crime; penis size, race, and crime (Rushton’s 1997 claims on penis size don’t ‘size up’ to the literature as I’ve shown two times); prostate-specific antigens, race, and prostate cancer; CAG repeats; intelligence and education and ‘intelligence’; and prenatal androgen exposure. His theory has large holes and doesn’t line up in some places, as he himself admits in his paper. He, as expected, cites Ross et al (1986) favorably in his analysis.
Testosterone can’t explain all of these differences, no matter if it’s prenatal androgen exposure or not, and a difference of 2.5 to 4.9 percent between blacks and whites regarding testosterone (Richard et al, 2014) won’t explain differences in crime, aggression, nor prostate cancer.
Other authors have attempted to also implicate testosterone as a major player in a wide range of evolutionary theories (Lynn, 1990; Rushton, 1997; Rushton, 1999; Hart, 2007; Rushton and Templer, 2012; Ellis, 2017). However, as can be seen by digging into this literature, these claims are not true and therefore we can discard the conclusions come to by the aforementioned authors since they’re based on false premises (testosterone being a cause for aggression, crime, and prostate cancer and r/K meaning anything to human races, it doesn’t)
Finally, to conclude this section, does testosterone explain racial differences in crime? No, racial differences in testosterone, however small, cannot be responsible for the crime gap between blacks and whites.
Testosterone and aggression
Testosterone and aggression, are they linked? Can testosterone tell us anything about individual differences in aggressive behavior? Surprisingly for most, the answer seems to be a resounding no. One example is the castration of males. Does it completely take away the urge to act aggressively? No, it does not. What is shown when sex offenders are castrated is that their levels of aggression decrease, but importantly, they do not decrease to 0. Robert Sapolsky writes on page 96 of his book Behave: The Biology of Humans at Our Best and Worst (2017) (pg 96):
… the more experience a male has being aggressive prior to castration, the more aggression continues afterward. In other words, the less his being aggressive in the future requires testosterone and the more it’s a function of social learning.
He also writes (pg 96-97):
On to the next issue that lessens the primacy of testosterone: What do individual levels of testosterone have to do with aggression? If one person higher testosterone levels than another, or higher levels this week than last, are they more likely to be aggressive?
Initially the answer seemed to be yes, as studies showed correlation between individual differences in testosterone levels and levels of aggression. In a typical study, higher testosterone levels would be observed in those male prisoners with higher rates of aggression. But being aggressive stimulates testosterone secretion; no wonder more aggressive individuals had higher levels. Such studies couldn’t disentangle chickens and eggs.
Thus, a better question is whether differences in testosterone levels among individuals predict who will be aggressive. And among birds, fish, mammals, and especially other primates, the answer is generally no. This has been studied extensively in humans, examining a variety of measures of aggression. And the answer is clear. To quote British endocrinologist John Archer in a definitive 2006 review, “There is a weak and inconsistent association between testosterone levels and aggression in [human] adults, and . . . administration of testosterone to volunteers typically does not increase aggression.” The brain doesn’t pay attention to testosterone levels within the normal range.
[…]
Thus, aggression is typically more about social learning than about testosterone, differing levels of testosterone generally can’t explain why some individuals are more aggressive than others.
Sapolsky also has a 1997 book of essays on human biology titled The Trouble With Testosterone: And Other Essays On The Biology Of The Human Predicament and he has a really good essay on testosterone titled Will Boys Just Be Boys? where he writes (pg 113 to 114):
Okay, suppose you note a correlation between levels of aggression and levels of testosterone among these normal males. This could be because (a) testosterone elevates aggression; (b) aggression elevates testosterone secretion; (c) neither causes the other. There’s a huge bias to assume option a while b is the answer. Study after study has shown that when you examine testosterone when males are first placed together in the social group, testosterone levels predict nothing about who is going to be aggressive. The subsequent behavioral differences drive the hormonal changes, not the other way around.
Because of a strong bias among certain scientists, it has taken do forever to convince them of this point.
[…]
As I said, it takes a lot of work to cure people of that physics envy, and to see interindividual differences in testosterone levels don’t predict subsequent differences in aggressive behavior among individuals. Similarly, fluctuations in testosterone within one individual over time do not predict subsequent changes in the levels of aggression in the one individual—get a hiccup in testosterone secretion one afternoon and that’s not when the guy goes postal.
And on page 115 writes:
You need some testosterone around for normal levels of aggressive behavior—zero levels after castration and down it usually goes; quadruple it (the sort of range generated in weight lifters abusing anabolic steroids), and aggression typically increases. But anywhere from roughly 20 percent of normal to twice normal and it’s all the same; the brain can’t distinguish among this wide range of basically normal values.
Weird…almost as if there is a wide range of ‘normal’ that is ‘built in’ to our homeodynamic physiology…
So here’s the point: differences in testosterone between individuals tell us nothing about individual differences in aggressive behavior; castration and replacement seems to show that, however broadly, testosterone is related to aggression “But that turns out to not be true either, and the implications of this are lost on most people the first thirty times you tell them about it. Which is why you’d better tell them about it thirty-one times, because it’s the most important part of this piece” (Sapolsky, 1997: 115).
Later in the essay, Sapolsky discusses a discusses 5 monkeys that were given time to form a hierarchy of 1 through 5. Number 3 can ‘throw his weight’ around with 4 and 5 but treads carefully around 1 and 2. He then states to take the third-ranking monkey and inject him with a ton of testosterone, and that when you check the behavioral data that he’d then be participating in more aggressive actions than before which would imply that the exogenous testosterone causes participation in more aggressive behavior. But it’s way more nuanced than that.
So even though small fluctuations in the levels of the hormone don’t seem to matter much, testosterone still causes aggression. But that would be wrong. Check out number 3 more closely. Is he now raining aggression and terror on any and all in the group, frothing in an androgenic glaze of indiscriminate violence. Not at all. He’s still judiciously kowtowing to numbers 1 and 2 but has simply become a total bastard to number 4 and 5. This is critical: testosterone isn’t causing aggression, it’s exaggerating the aggression that’s already there.
The correlation between testosterone and aggression is between .08 and .14 (Book, Starzyk, and Quinsey, 2001; Archer, Graham-Kevan, and Davies, 2005; Book and Quinsey, 2005). Therefore, along with all of the other evidence provided in this article, it seems that testosterone and aggression have a weak positive correlation, which buttresses the point that aggression concurrent increases in testosterone.
Sapolsky then goes on to discuss the amygdala’s role in fear processing. The amygdala has its influence on aggressive behavior through the stria terminalis, which is a bunch of neuronal connections. How the amygdala influences aggression is simple: bursts of electrical excitation called action potentials go up and down the stria terminalis which changes the hypothalamus. You can then inject testosterone right into the brain and will it cause the same action potentials that surge down the stria terminalis? No, it does not turn on the pathway at all. This only occurs only if the amygdala is already sending aggression-provoking action potentials down the stria terminalis with testosterone increasing the rate of action potentials you’re shortening the rest time between them. So it doesn’t turn on this pathway, it exaggerates the preexisting pattern, which is to say, it’s exaggerating the response to environmental triggers of what caused the amygdala to get excited in the first place.
He ends this essay writing (pg 119):
Testosterone is never going to tell us much about the suburban teenager who, in his after-school chess club, has developed a particularly aggressive style with his bishops. And it certainly isn’t going to tell us much about the teenager in some inner-city hellhole who has taken to mugging people. “Testosterone equals aggression” is inadequate for those who would offer a simple solution to the violent male—just decrease levels of those pesky steroids. And “testosterone equals aggression” is certainly inadequate for those who would offer a simple excuse: Boys will be boys and certain things in nature are inevitable. Violence is more complex than a single hormone. This is endocrinology for the bleeding heart liberal—our behavioral biology is usually meaningless outside of the context of social factors and the environment in which it occurs.
Injecting individuals with supraphysiological doses of testosterone as high as 200 and 600 mg per week does not cause heightened anger or aggression (Tricker et al, 1996; O’Connor et, 2002). This, too, is a large blow for the testosterone-induces-aggression hypothesis. Because aggressive behavior heightens testosterone, testosterone doesn’t heighten aggressive behavior. (This is the causality that has been looked for, and here it is. The causality is not in the other direction.) This tells us that we need to be put into situations for our aggression to rise and along with it, testosterone. I don’t even see how people could think that testosterone could cause aggression. It’s obvious that the environmental trigger needs to be there first in order for the body’s physiology to begin testosterone production in order to prepare for the stimulus that caused the heightened testosterone production. Once the trigger occurs, then it can and does stay heightened, especially in areas where dominance contests would be more likely to occur, which would be low-income areas (Mazur, 2006, 2016).
(Also read my response to Batrinos, 2012, my musings on testosterone and race, and my responses to Robert Lindsay and Sean Last.)
Lastly, one thing that gets on my nerves that people point to to attempt to show that testosterone and its derivatives cause violence, aggression etc is the myth of “roid rage” which is when an individual objects himself with testosterone, anabolic steroids or another banned substance, and then the individual becomes more aggressive as a result of more free-flowing testosterone in their bloodstream.
The problem here is that people believe what they hear on the media about steroids and testosterone, and they’re largely not true. One large analysis was done to see the effects of steroids and other illicit drug use on behavior, and what was found was that after controlling for other substance use “Our results suggest that it was not lifetime steroid use per se, but rather co-occurrring polysubstance abuse that most parsimoniously explains the relatively strong association of steroid use and interpersonal violence” (Lundholm et al, 2015). So after controlling for other drugs used, men who use steroids do not go to prison and be convicted of violence after other polysubstance use was controlled for, implying that is what’s driving interpersonal violence, not the substance abuse of steroids.
Conclusion
Numerous myths about testosterone have been propagated over the decades, which are still believed in the new millennium despite numerous other studies and arguments to the contrary. As can be seen, the myths that people believe about testosterone are easily debunked. Numerous papers (with better methodology than Ross et al) attest to the fact that testosterone levels aren’t as high as was believed decades ago between the races. Diet can explain a lot of the variation, especially vitamin D intake. Injecting men with supraphysiological doses of testosterone does not heighten anger nor aggression. It does not even heighten prostate cancer severity.
Racial differences in testosterone are also not as high as people would like to believe, there is even an opposite relationship with Asians having higher levels and whites having lower (which wouldn’t, on average, imply femininity) testosterone levels. So as can be seen, the attempted r/K explanations from Rushton et al don’t work out here. They’re just outright wrong on testosterone, as I’ve been arguing for a long while on this blog.
Testosterone doesn’t cause aggression, aggression causes heightened testosterone. It can be seen from studies of men who have been castrated that the more crime they committed before castration, the more crime they will commit after which implies a large effect of social learning on violent behavior. Either way, the alarmist attitudes of people regarding testosterone, as I have argued, are not needed because they’re largely myths.
Is Diet An IQ Test?
1350 words
Dr. James Thompson is a big proponent of ‘diet being an IQ test‘ and has written quite a few articles on this matter. Though, the one he published today is perhaps the most misinformed.
He first shortly discusses the fact that 200 kcal drinks are being marketed as ‘cures’ for type II diabetes. People ‘beat’ the disease with only 200 kcal drinks. Sure, they lost weight, lost their disease. Now what? Continue drinking the drinks or now go back to old dietary habits? Type II diabetes is a lifestyle disease, and so can be ameliorated with lifestyle interventions. Though, Big Pharma wants you to believe that you can only overcome the disease with their medicines and ‘treatments’ along with the injection of insulin from your primary care doctor. Though, this would only exacerbate the disease, not cure it. The fact of the matter is this: these ‘treatments’ only ‘cure’ the proximate causes. The ULTIMATE CAUSES are left alone and this is why people fall back into habits.
When speaking about diabetes and obesity, this is a very important distinction to make. Most doctors, when treating diabetics, only treat the proximate causes (weight, symptoms that come with weight, etc) but they never get to the root of the problem. The root of the problem is, of course, insulin. The main root is never taken care of, only the proximate causes are ‘cured’ through interventions, however, the underlying cause of diabetes, and obesity as well is not taken care of because of doctors. This, then, leads to a neverending cycle of people losing a few pounds or whatnot and then they, expectedly, gain it back and they have to re-do the regimen all over again. The patient never gets cured, Big Pharma, hospitals et al get to make money off not curing a patients illness by only treating proximate and not ultimate causes.
Dr. Thompson then talks about a drink for anorexics, called ‘Complan“, and that he and another researcher gave this drink to anorexics, giving them about 3000 kcals per day of the drink, which was full of carbs, fat and vitamins and minerals (Bhanji and Thompson, 1974).
The total daily calorific intake was 2000-3000 calories, resulting in a mean weight gain of 12.39 kilos over 53 days, a daily gain of 234 grams, or 1.64 kilos (3.6 pounds) a week. That is in fact a reasonable estimate of the weight gains made by a totally sedentary person who eats a 3000 calorie diet. For a higher amount of calories, adjust upwards. Thermodynamics.
Thermodynamics? Take the first law. The first law of thermodynamics is irrelevant to human physiology (Taubes, 2007; Taubes, 2011; Fung, 2016). (Also watch Gary Taubes explain the laws of thermodynamics.) Now take the second law of thermodynamics which “states that the total entropy can never decrease over time for an isolated system, that is, a system in which neither energy nor matter can enter nor leave.” People may say that ‘a calorie is a calorie’ therefore it doesn’t matter whether all of your calories come from, say, sugar or a balanced high fat low carb diet, all weight gain or loss will be the same. Here’s the thing about that: it is fallacious. Stating that ‘a calorie is a calorie’ violates the second law of thermodynamics (Feinman and Fine, 2004). They write:
The second law of thermodynamics says that variation of efficiency for different metabolic pathways is to be expected. Thus, ironically the dictum that a “calorie is a calorie” violates the second law of thermodynamics, as a matter of principle.
So talk of thermodynamics when talking about the human physiological system does not make sense.
He then cites a new paper from Lean et al (2017) on weight management and type II diabetes. The authors write that “Type 2 diabetes is a chronic disorder that requires lifelong treatment. We aimed to assess whether intensive weight management within routine primary care would achieve remission of type 2 diabetes.” To which Dr. Thompson asks ‘How does one catch this illness?” and ‘Is there some vaccination against this “chronic disorder”?‘ The answer to how does one ‘catch this illness’ is simple: the overconsumption of processed carbohydrates, constantly spiking insulin which leads to insulin resistance which then leads to the production of more insulin since the body is resistant which then causes a vicious cycle and eventually insulin resistance occurs along with type II diabetes.
Dr. Thompson writes:
Patients had been put on Complan, or its equivalent, to break them from the bad habits of their habitual fattening diet. This is good news, and I am in favour of it. What irritates me is the evasion contained in this story, in that it does not mention that the “illness” of type 2 diabetes is merely a consequence of eating too much and becoming fat. What should the headline have been?
Trial shows that fat people who eat less become slimmer and healthier.
I hope this wonder treatment receives lots of publicity. If you wish to avoid hurting anyone’s feelings just don’t mention fatness. In extremis, you may talk about body fat around vital organs, but keep it brief, and generally evasive.
So you ‘break bad habits’ by introducing new bad habits? It’s not sustainable to drink these low kcal drinks and expect to be healthy. I hope this ‘wonder treatment’ does not receive a lot of publicity because it’s bullshit that will just line the pockets of Big Pharma et al, while making people sicker and, the ultimate goal, having them ‘need’ Big Pharma to care for their illness—when they can just as easily care for it themselves.
‘Trial shows that fat people who eat less become slimmer and healthier’. Or how about this? Fat people that eat well and exercise, up to 35 BMI, have no higher risk of early death then someone with a normal BMI who eats well and exercises (Barry et al, 2014). Neuroscientist Dr. Sandra Aamodt also compiles a wealth of solid information on this subject in her 2016 book “Why Diets Make Us Fat: The Unintended Consequences of Our Obsession with Weight Loss“.
Dr. Thompson writes:
I see little need to update the broad conclusion: if you want to lose weight you should eat less.
This is horrible advice. Most diets fail, and they fail because the ‘cures’ (eat less, move more; Caloric Reduction as Primary: CRaP) are garbage and don’t take human physiology into account. If you want to lose weight and put your diabetes into remission, then you must eat a low-carb (low carb or ketogenic, doesn’t matter) diet (Westman et al, 2008; Azar, Beydoun, and Albadri, 2016; Noakes and Windt, 2016; Saslow et al, 2017). Combine this with an intermittent fasting plan as pushed by Dr. Jason Fung, and you have a recipe to beat diabesity (diabetes and obesity) that does not involve lining the pockets of Big Pharma, nor does it involve one sacrificing their health for ‘quick-fix’ diet plans that never work.
In sum, diets are not ‘IQ tests’. Low kcal ‘drinks’ to ‘change habits’ of type II diabetics will eventually exacerbate the problem because when the body is in extended caloric restriction, the brain panics and releases hormones to stimulate appetite while stopping hormones that cause you to be sated and stop eating. This is reality; these studies that show that eating or drinking 800 kcal per day or whatnot are based on huge flaws: the fact that this could be sustainable for a large number of the population is not true. In fact, no matter how much ‘willpower’ you have, you will eventually give in because willpower is a finite resource (Mann, 2014).
There are easier ways to lose weight and combat diabetes, and it doesn’t involve handing money over to Big Pharma/Big Food. You only need to intermittently fast, you’ll lose weight and your diabetes will not be a problem, you’ll be able to lose weight and will not have problems with diabetes any longer (Fung, 2016). Most of these papers coming out recently on this disease are garbage. Real interventions exist, they’re easier and you don’t need to line the pockets of corporations to ‘get cured’ (which never happens, they don’t want to cure you!)
Black-White Differences in Physiology
2050 words
Black-white differences in physiology can tell a lot about how the two groups have evolved over time. On traits like resting metabolic rate (RMR), basal metabolic rate (BMR), adiposity, heart rate, Vo2 max, etc. These differences in physiological variables between groups, then, explain part of the reason why there are different outcomes in terms of life quality/mortality between the two groups.
Right away, by looking at the average black and average white, you can see that there are differences in somatype. So if there are differences in somatype, then there must be differences in physiological variables, and so, this may be a part of the cause of, say, differing obesity rates between black and white women (Albu et al, 1997) and even PCOS (Wang and Alvero, 2013).
Resting metabolic rate
Resting metabolic rate is your body’s metabolism at rest, and is the largest component of the daily energy budget in modern human societies (Speakman and Selman, 2003). So if two groups, on average, differ in RMR, then one with the lower RMR may have a higher risk of obesity than the group with the higher RMR. And this is what we see.
Black women do, without a shadow of a doubt, have a lower BMR, lower PAEE (physical activity energy expenditure) and TDEE (total daily expenditure) (Gannon, DiPietro, and Poehlman, 2000). Knowing this, then it is not surprising to learn that black women are also the most obese demographic in the United States. This could partly explain why black women have such a hard time losing weight. Metabolic differences between ethnic groups in America—despite living in similar environments—show that a genetic component is responsible for this.
There are even predictors of obesity in post-menopausal black and white women (Nicklas et al, 1999). They controlled for age, body weight and body composition (variables that would influence the results—no one tell me that “They shouldn’t have controlled for those because it’s a racial confound!”) and found that despite having a similar waist-to-hip ratio (WHR) and subcutaneous fat area, black women had lower visceral fat than white women, while fasting glucose, insulin levels, and resting blood pressure did not differ between the groups. White women also had a higher Vo2 max, which remained when lean mass was controlled for. White women could also oxidize fat at a higher rate than black women (15.4 g/day, which is 17% higher than black women). When this is expressed as percent of total kcal burned in a resting state, white women burned more fat than black women (50% vs 43%). I will cover the cause for this later in the article (one physiologic variable is a large cause of these differences).
We even see this in black American men with more African ancestry—they’re less likely to be obese (Klimentidis et al 2016). This, too, goes back to metabolic rate. Black American men have lower levels of body fat than white men (Vickery et al, 1988; Wagner and Heyward, 2000). All in all, there are specific genetic variants and physiologic effects, which cause West African men to have lower central (abdominal) adiposity than European men and black women who live in the same environment as black men—implying that genetic and physiologic differences between the sexes are the cause for this disparity. Whatever the case may be, it’s interesting and more studies need to be taken out so we can see how whatever gene variants are *identified* as protecting against central adiposity work in concert with the system to produce the protective effect. Black American men have lower body fat, therefore they would have, in theory, a higher metabolic rate and be less likely to be obese—while black women have the reverse compared to white women—a lower metabolic rate.
Skeletal muscle fiber
Skeletal muscle fibers are the how and why of black domination in explosive sports. This is something I’ve covered in depth. Type II fibers contract faster than type I. This has important implications for certain diseases that black men are more susceptible to. Though the continuous contraction of the fibers during physical activity leads to a higher disease susceptibility in black men—but not white men (Tanner et al, 2001). If you’re aware of fiber type differences between the races (Ama et al, 1986; Entine, 2000; Caeser and Henry, 2015); though see Kerr (2010’s) article The Myth of Racial Superiority in Sports for another view. That will be covered here in the future.
Nevertheless, fiber typing explains racial differences in sports, with somatype being another important variable in explaining racial disparities in sports. Two main variables that work in concert are the somatype (pretty much body measurements, length) and the fiber type. This explains why blacks dominate baseball and football; this explains why ‘white men can’t jump and black men can’t swim’. Physiological variables—not only ‘motivation’ or whatever else people who deny these innate differences say—largely explain why there are huge disparities in these sports. Physiology is important to our understanding of how and why certain groups dominate certain sports.
This is further compounded by differing African ethnies excelling in different running sports depending on where their ancestors evolved. Kenyans have an abundance of type I fibers whereas West Africans have an abundance of type II fibers. (Genetically speaking, ‘Jamaicans’ don’t exist; genetic testing shows them to come from a few different West African countries.) Lower body symmetry—knees and ankles—show that they’re more symmetrical than age-matched controls (Trivers et al, 2014). This also goes to show that you can’t teach speed (Lombardo and Deander, 2014). Though, of course, training and the will to want to do your best matter as well—you just cannot excel in these competitions without first and foremost having the right physiologic and genetic make-up.
Further, although it’s only one gene variant, ACTN3 and ACE explain a substantial percentage of sprint time variance, which could be the difference between breaking a world record and making a final (Papadimitriou et al, 2016). So, clearly, certain genetic variants matter more than others—and the two best studied are ACTN3 and ACE. Some authors, though, may deny the contribution of ACTN3 to elite athletic performance—like one researcher who has written numerous papers on ACTN3, Daniel MacArthur. However, elite sprinters are more likely to carry the RR ACTN3 genotype compared to the XX ACTN3 genotype, and the RR ACTN3 genotype—when combined with type II fibers and morphology—lead to increased athletic performance (Broos et al, 2016). It’s also worth noting that 2 percent of Jamaicans carry the XX ACTN3 genotype (Scott et al, 2010), so this is another well-studied variable that lends to superior running performance in Jamaicans.
In regards to Kenyans, of course when you are talking about genetic reasons for performance, some people don’t like it. Some may say that certain countries dominate in X, and that for instance, North Africa is starting to churn out elite athletes, should we begin looking for genetic advantages that they possess (Hamilton, 2000)? Though people like Hamilton are a minority view in this field, I have read a few papers that there is no evidence that Kenyans possess a pulmonary system that infers a physiologic advantage over whites (Larsen and Sheel, 2015).
People like these three authors, however, are in the minority here and there is a robust amount of research that attests to East African running dominance being genetic/physiologic in nature—though you can’t discredit SES and other motivating variables (Tucker, Onywera, and Santos-Concejero, 2015). Of course, a complex interaction between SES, genes, and environment are the cause of the success of the Kalenjin people of Kenya, because they live and train in such high altitudes (Larsen, 2003), though the venerable Bengt Saltin states that the higher Vo2 max in Kenyan boys is due to higher physical activity during childhood (Saltin et al, 1995).
Blood pressure
The last variable I will focus on (I will cover more in the future) is blood pressure. It’s well known that blacks have higher blood pressure than whites—with black women having a higher BP than all groups—which then leads to other health implications. Some reasons for the cause are high sodium intake in blacks (Jones and Hall, 2006); salt (Lackland, 2014; blacks had a similar sensitivity than whites, but had a higher blood pressure increase); while race and ethnicity was a single independent predictor of hypertension (Holmes et al, 2013). Put simply, when it comes to BP, ethnicity matters (Lane and Lip, 2001).
While genetic factors are important in showing how and why certain ethnies have higher BP than others, social factors are arguably more important (Williams, 1992). He cites stress, socioecologic stress, social support, coping patterns, health behavior, sodium, calcium, and potassium consumption, alcohol consumption, and obesity. SES factors, of course, lead to higher rates of obesity (Sobal and Stunkard, 1989; Franklin et al, 2015). So, of course, environmental/social factors have an effect on BP—no matter if the discrimination or whatnot is imagined by the one who is supposedly discriminated against, this still causes physiologic changes in the body which then lead to higher rates of BP in certain populations.
Poverty does affect a whole slew of variables, but what I’m worried about here is its effect on blood pressure. People who are in poverty can only afford certain foods, which would then cause certain physiologic variables to increase, exacerbating the problem (Gupta, de Wit, and McKeown, 2007). Whereas diets high in protein predicted lower BP in adults (Beundia et al, 2015). So this is good evidence that the diets of blacks in America do increase BP, since they eat high amounts of salt, low protein and high carb diets.
Still, others argue that differences in BP between blacks and whites may not be explained by ancestry, but by differences in education, rather than genetic factors (Non, Gravlee, and Mulligan, 2012). Their study suggests that educating black Americans on the dangers and preventative measures of high BP will reduce BP disparities between the races. This is in-line with Williams (1992) in that the social environment is the cause for the higher rates of BP. One hypothesis explored to explain why this effect with education was greater in blacks than whites was that BP-related factors, such as stress, poverty and racial discrimination (remember, even if no racial discrimination occurs, any so-called discrimination is in the eye of the beholder so that will contribute to a rise in physiologic variables) and maybe social isolation may be causes for this phenomenon. Future studies also must show how higher education causes lower BP, or if it only serves as other markers for the social environment. Nevertheless, this is an important study in our understanding of how and why the races differ in BP and it will go far to increase our understanding of this malady.
Conclusion
This is not an exhaustive list—I could continue writing about other variables—but these three are some of the most important as they are a cause for higher mortality rates in America. Understanding the hows and whys of these variables will have us better equipped to help those who suffer from diseases brought on by these differences in physiological factors.
The cause for some of these physiologic differences come down to evolution, but still others may come down to the immediate obesogenic environment (Lake and Townshend, 2006) which is compounded by lower SES. Since high carbs diets increase BP, this explains part of the reason why blacks have higher BP, along with social and genetic factors. Muscle fiber typing is set by the second trimester, and no change is seen after age 6 (Bell, 1980). Resting metabolic rate gap differences between black and white women can be closed, but not completely, if black women were to engage in exercise that use their higher amounts of type II muscle fibers (Tanner et al, 2001). This research is important to understand differences in racial mortality; because when we understand them then we can begin to theorize on how and why we see these disparities.
Physiologic differences between the races are interesting, they’re easily measurable and they explain both disparities in sports and mortality by different diseases. Once we study these variables more, we will be better able to help people with these variables—race be dammed. Race is a predictor here, only because race is correlated with other variables that lead to negative health outcomes. So once we understand how and why these differences occur, then we can help others with similar problems—no matter their race.
No, Soy Doesn’t Feminize Males
1400 words
There are a few things I constantly see around the Internet that really irk me. One of those things is the meme that soy causes feminization in males. Some people may see a picture of a guy with soft facial features and they’ll say ‘oh that’s a soy boy.’ They do not show a good understanding of soy and its chemicals. As usual, overblown claims from people who have no specialty in what they’re talking about have an answer—just one they do not want to hear.
I have no idea when this craze of blaming soy and other foods on ‘feminization of males’ (I have done so a few years ago, though I realize my error now), but it’s starting to get out of hand. I see this around Twitter a lot; ‘oh it’s the soy making men beta’ or ‘they know exactly what soy does to men and they still push it!’. The whole scare about soy is that soy has phytoestrogens called isoflavones, and these phytoestrogens then may mimic estrogen production in the body. (It’s worth noting that phytoestrogens do not always mimic estrogens, despite some people’s protesting to me that “It has estrogen right in the name!”.)
Many men have blamed soy and phytoestrogens/isoflavones on their growing breasts, having a flaccid penis, slow beard growth (that’d be enough motivation for me to do something about it, to be honest), loss of hair on his arms and legs, and, he says “Men aren’t supposed to have breasts … It was like my body is feminizing.” So this man went to an endocrinologist and was told that he had gynecomastia—enlarged breasts in men. He got tested for a whole slew of maladies, but they all came back negative. Then the doctor told him to be very specific with his diet. The man then said that he was lactose intolerant and that he drank soy milk instead of cow’s milk. The doctor then asked him how much soy milk he drank per day, to which the man responded 3 quarts. Was soy the cause of his gynecomastia and other negative health effects? I don’t think so.
For men and boys, soy does not seem to have an effect on estrogen and other hormone levels, nor does it seem to have an effect on development and fertility:
A handful of individuals and organizations have taken an anti-soy position and have questioned the safety of soy products. In general, this position latches to statistically insignificant findings, understates how powerfully the research refutes many of the main anti-soy points, and relies heavily on animal research studies, which are medically irrelevant to human health.
Most of those people are ‘activists’ who do not understand the intricacies of what they are talking about. In the field of nutrition, it’s dumb to take studies done on animals nd then extrapolate that data to humans. It does not make sense.
There are reasons not to eat soy that do not go along with this dumb hysteria from people who do not know what they are talking about. One reasons is that, when compared to casein protein, soy protein is inferior (Luiking et al, 2005). Further, data is “inconsistent or inadequate in supporting most of the suggested health benefits of consuming soy protein or ISF” (Xiao, 2008). People may cite studies like Lephart et al (2002) where they write in their article Neurobehavioral effects of dietary soy estrogens:
Whether these observations hold true for the human brain remains to be established and will only become apparent from long-term clinical studies. One point that needs clarification relates to the timing of brain development in the rat, which differs from that in the human. (pg. 12)
Rats and humans are different; I don’t think I need to say that. Though people who cite these studies (uncritically) as ‘evidence’ that soy lowers T, causes infertility problems, and feminization just look to grab anything that affirms their fearmongering views.
Jargin (2014) writes in his article Soy and phytoestrogens: possible side effects:
Feminizing effect of phytoestrogens and soy products may be subtle, detectable only statistically in large populations; it can be of particular importance for children and adolescents. This matter should be clarified by independent research, which can have implications for the future of soy in the agriculture.
If it’s only identifiable in statistically large populations, then it is meaningless. Though, East Asians seem to be adapted to soy and so (like all studies should note), these results should not be extrapolated to other races/ethnies (Jargin, 2014). I do agree with that assessment; I do not agree with the claim that these phytoestrogens would cause problems that would only be seen in statistically large populations.
If people want to use rat studies to show that soy supposedly raises estrogen levels, then I will use a rat study showing the opposite (Song, Hendrich, and Murphy, 1999). They showed that the effects of the isoflavones from soy were only weakly estrogenic. Further, Mitchell et al (2001) write in their article Effect of a phytoestrogen food supplement on reproductive health in normal males:
The phytoestrogen supplement increased plasma genistein and daidzein
concentrations to approx. 1 µM and 0.5 µM respectively; yet, there was no observable effect on endocrine measurements, testicular volume or semen parameters over the study period. This is the first study to examine the effects of a phytoestrogen supplement on reproductive health in males. We conclude that the phytoestrogen dose consumed had no effect on semen quality.
The men in this study took a daily isoflavone supplement containing 40 mg of isoflavone every day for two months and still did not show negative effects. Looking at rat studies and then extrapolating that to humans doesn’t make sense.
Finally, there was a large meta-analysis by Messina (2010) who writes:
The intervention data indicate that isoflavones do not exert feminizing effects on men at intake levels equal to and even considerably higher than are typical for Asian males.
And from the conclusion:
Isoflavone exposure at levels even greatly exceeding reasonable dietary intakes does not affect blood T or estrogen levels in men or sperm and semen parameters. The ED-related findings in rats can be attributed to excessive isoflavone exposure and to differences in isoflavone metabolism between rodents and humans. Thus, men can feel confident that making soy a part of their diet will not compromise their virility or reproductive health.
Now, I know that proponents of the hypothesis of soy feminizing males would say to me “Why don’t you just eat a bunch of soy and see what happens then, if it is fine for you?” Well, unlike most people, I eat a strict diet and soy is not part of it.
Soy isoflavones are currently being researched in regards to the prevention and treatment of diabetes, cardiovascular disease, cancer, osteoporosis, and neuroprotection (Kalaiselven et al, 2010), while the nutritional and health benefits of soy isoflavones are currently being studied (Friedman and Brandon, 2001; McCue and Shetty, 2004). Soy foods may also be optimal for bone health (Lanou, 2011).
In conclusion, as you can see, the claims of soy causing feminizing effects on men are largely overblown. People extrapolate data from rat studies to humans, which doesn’t make any sense. To paraphrase Dr. Jason Fung, imagine 2 lions are watching a deer eat. They see how strong and in shape the deer are eating grass. So the lions, noticing that the deer are healthy eating grass, begin to eat grass too thinking it is healthy and they die. One hundred years later, 2 deer are watching a lion eat meat and sees how strong, fast, and healthy it is. They then draw the conclusion that eating meat is healthy and will do the same for them and they eat meat and die. The point of the analogy is that just because studies on animals show X and therefore Y, that does not mean that it will hold for humans! This is something very simple for people to understand, though most do not.
Most people search for things to prove their assumptions without having an actual understanding of the biological mechanisms of what they’re talking about. People should learn some of the literature before they cite studies that supposedly back their biases, because they would then see that it is not as nuanced as they believe.
(Note: Diet changing under Doctor’s supervision only.)
