As is the case with the other lines of evidence that intend to provide sociobiological evidence in support of the genetic basis of human behavior and development (relating to homology, heritability, and adaptation), Rushton’s work reduces to no evidence at all. (Lerner, 2018)
Introduction
From 1985 until his death in 2012, J. P. Rushton attempted to marshal all of the data and support he could for a theory called r-K selection theory or Differential K theory (Rushton, 1985). The theory posited that while humans were the most K species of all, some human races were more K than others, so it then followed that some human races were more r than others. Rushton then collated mass amounts of data and wrote what would become his magnum opus, Race, Evolution and Behavior (Rushton, 1997). So in the r/K theory first proposed by MacArthur and Wilson, unstable, unpredictable environments favored an r strategy whereas a stable, predictable environments favored a K strategy. (See here for my response to Rushton’s r/K.)
So knowing this, one of the suite of traits Rushton put on his r/K matrix was twinning rates. Rushton (1997: 6) stated:
the rate of dizygotic twinning, a direct index of egg production, is less than 4 per 1,000 births among Mongoloids, 8 per 1,000 among Caucasoids, and 16 or greater per 1,000 among Negroids.
I won’t contest the claim that the rate in DZ twinning is higher by race—because it’s pretty well-established with recent data that blacks are more likely to have twins than whites (that is, blacks have a slightly higher chance of having twins than whites, who have a slightly higher chance of having twins than Asians) (Santana, Surita, and Cecatti, 2018; Wang, Dongarwar, and Salihu, 2020; Monden, Pison, and Smits, 2021)—I’m merely going to contest the causes of DZ twinning. Because it’s clear that Rushton was presuming this to be a deeply evolutionary trait since a highs rate of twins—in an evolutionary context—would mean that there would be a higher chance for children of a particular family to survive and therefore spread their genes and thusly would, in his eyes, lend credence to his claim that Africans were more r compared to whites who were more r compared to Asians.
But to the best of my knowledge, Rushton didn’t explain why, biologically, blacks would have more twins than whites—he merely said “This race has more twins than this race, so this lends credence to my theory.” That is, he didn’t posit a biological mechanism that would instantiate a higher rate of twinning in blacks compared to whites and Asians and then explain how environmental effects wouldn’t have any say in the rate of twinning between the races. However, I am privy to environmental factors that would lead to higher rates of twinning and I am also privy to the mechanisms of action that allow twinning to occur (eg phytoestrogens, FSH, LH, and IGF). And while these are of course biological factors, I will show that there are considerable effects of environmental interactions like diet on the levels of these hormones which are associated with twinning. I will also explain how these hormones are related to twinning.
While the claim that there is a difference in rate of DZ twinning by race seems to be true, I don’t think it’s a biological trait, nevermind an evolutionary one as Rushton proposed (because even if Rushton’s r/K were valid, “Negroids” would be K and “Mongoloids” would be r, Anderson, 1991). Nonetheless, Rushton’s r/K theory is long-refuted, though he did call attention to some interesting observations (which other researchers never ignored, they just didn’t attempt some grand theory of racial differences).
Follicle stimulating hormone, leutinizing hormone, and insulin-like growth factor
We know that older women are more likely to have twins while younger women are less likely (Oleszczuk et al, 2001), so maternal age is a factor. As women age, a hormone called follicle stimulating hormone (FSH) increases due to a decline in estrogen, and it is one of the earliest signs of female reproductive aging (McTavish et al, 2007), being one of the main biomarkers of ovarian reserve tested on day 3 of the menstrual cycle (Roudebush, Kivens, and Mattke, 2008). It is well established that twinning is different in different geographic locations, that the rate of MZ twins is constant at around 3.5 to 4 per 1,000 births (so what is driving the differences is the birth of DZ twins), and that it increases due to an increase in FSH (Santana, Surita, and Cecatti, 2018). We also know that pre-menopausal women who have given birth to DZ twins have higher levels of FSH on the third day of their menstrual cycle (Lambalk et al, 1998).
So if FSH levels stay too high for too long then multiple eggs are released, which could lead to an increase in DZ twinning. FSH stimulates the maturation and growth of ovarian follicles, each of which contains an immature egg called an oocyte. FSH acts on the ovaries to promote the development of multiple ovarian follicles during pregnancy, a process which is called recruitment. In a normal menstrual cycle, only one follicle is stimulated to release one egg; but when FSH levels are elevated, this results in the development and maturation of more than one follicle which is known as polyovulation. Polyovulation then increases the chance of the release of multiple eggs during ovulation. Thus, if more than one egg is released during a menstrual cycle, and they both are fertilized, it can then lead to the development of DZ twins.
Along with FSH, we also have luetenizing hormone (LH). So FSH and LH act synergistically (Raju et al, 2013). LH, like FSH, isn’t directly responsible for the increase in twinning, but the process that it allows (playing a role in ovulation) is a crucial factor in twinning. So LH is responsible for triggering ovulation, which is the release of a mature egg from the ovarian follicle. (Ovulation occurs typically 24 to 36 hours after LH increases.) In a typical menstrual cycle, only one follicle is stimulated to release one egg, which is triggered by the surge in LH. But if there are multiple mature follies in the ovaries (which could be influenced by FSH), then a surge in LH can lead to the release of more than one egg. So the interaction of LH with other hormone like FSH, along with the presence of multiple mature follicles, can be associated with having a higher chance of having DZ twins. FSH therapies are also used in assisted reproduction (eg Munoz et al, 1995 in mice; Ferraretti et al, 2004; Pang, 2005; Pouwer, Farquhar, and Kremer, 2015; Fatemi et al, 2021).
So when it comes to FSH, we know that malnutrition may play a role in twinning, and also that wild yams—a staple food in Nigeria—increases phytoestrogens which increase FSH in the body of women (Bartolus, et al, 1999). Wild yams have been used to increase estrogen in women’s bodies (due to the phytoestrogens they contain), and it enhances estradiol through the mechanism of binding to estrogen receptor sites (Hywood, 2008). And since Nigeria has the highest rate of twinning in the world (Santana, Surita, and Cecatti, 2018), and their diet is wild yam-heavy (Bartolus, et al, 1999), it seems that this fact would go a long way in explaining why they have higher rates of twinning. Mount Sinai says that “Although it does not seem to act like a hormone in the body, there is a slight risk that wild yam could produce similar effects to estrogen.” It acts as a weak phytoestrogen (Park et al, 2009). (But see Beckham, 2002.) But when phytoestrogens are consumed, they can then bind to estrogen receptors in the body and trigger estrogenic effects which could then lead to the potential stimulation and release of multiple eggs which would increase the chance of DZ twinning.
One study showed that black women, in comparison to white women, had “lower follicular phase LH:FSH ratios” (Reuttman et al, 2002; cf Marsh et al, 2011), while Randolph et al (2004) showed that black women had higher FSH than Asian and white women. So the lower LH:FSH ratio could affect the timing and regulation of ovulation, and a lower LH:FSH level could reduce the chances of premature ovulation and could affect the release of multiple eggs.
Lastly, when it comes to insulin-like growth factor (IGF), this could be influenced by a high protein diet or a high carb diet. Diets high in high glycemic carbs can lead to increase insulin production which would then lead to increased IGF levels. Just like with FSH and LH, increased levels of IGF could also in concert with the other two hormones influence the maturation and release of multiple eggs during a menstrual cycle which would then increase the chance of twinning (Yoshimura, 1998). IGF can also stimulate the growth and development of multiple follicles (Stubbs et al, 2013) and have them mature early if IGF levels are high enough (Mazerbourgh and Monget, 2018). This could then also lead to polyovulation, triggering the release of more than one egg during ovulation. IGF can also influence the sensitivity of the ovaries to hormonal signals, like those from the pituitary gland, which then leads to enhanced ovarian sensitivity to hormones like FSH and LH which then, of course, would act synergistically increasing the rate of dizygotic twinning. (See Mazerbourgh and Monget, 2018 for a review of this.)
So we know that black women have higher levels of IGF-1 and free IGF-1—but lower IGF-2 and IGFBP-3—than white women (Berrigan et al, 2010; Fowke et al, 2011). The higher IGF-1 levels in black women could lead to increase ovarian sensitivity to FSH and LH, and thus enhanced ovarian sensitivity could lead to the promotion and release of multiple eggs during ovulation. The lower IGF-2 levels could contribute to the balance of IGF-1 and IGF-2, which would then further influence the ovarian sensitivity to other hormones. IGFBP-3 is a binding protein which regulated the bioavailability of IGF-1, so lower levels of IGFBP-3 could lead to higher concentrations of free IGF-1, which would then further stimulate the ovarian follicles and could lead to polyovulation, leading to increased twinning. Though there is some evidence that this difference does have a “genetic basis” (Higgins et al, 2005), we know that dietary factors do have an effect on IGF levels (Heald et al, 2003).
Rushton’s misinterpretations
Rushton got a ton wrong, but he was right about some things too (which is to be expected if you’re looking to create some grand theory of racial differences). I’m not too worried about that. But what I AM worried about, is Rushton’s outright refusal to address his most serious critics in the literature, most importantly Anderson (1991) and Graves (2002 a, b). If you check his book (Rushton, 1997: 246-248), his responses are hardly sufficient to address the devestating critiques of his theory. (Note how Rushton never responded to Graves, 2002—ever.) Gorey and Cryns (1995) showed how Rushton cherry-picked what he liked for his theory while stating that “any behavioral differences which do exist between blacks, whites and Asian Americans for example, can be explained in toto by environmental differences which exist between them” while Ember, Ember, and Peregrine (2003) concluded similarly. (Rushton did respond to Gorey and Cryns, but not Ember, Ember, and Peregrine.) Cernovsky and Littman (2019) also showed how Rushton cherry-picked his INTERPOL crime data.
Now that I have set the stage for Rushton’s “great” scholarship, let’s talk about the response he got to his twinning theory.
Allen et al (1992) have a masterful critique of Rushton’s twinning theory. They review twinning stats in other countries across different time periods and come to conclude that “With such a wide overlap between races, and such great variation within races, twinning rate is probably no better than intelligence as an index of genetic status for racial groups.” They also showed that the twinning mechanism didn’t seem to be a relevant factor in survival, until the modern day with the advancement of our medical technologies, that is. So since twinning increases the risk for death in the mother (Steer, 2007; Santana et al, 2018). Rushton also misinterpreted numerous traits associated with twinning:
individual twin proneness and its correlates do not provide Rushton’s desired picture of a many-faceted r- strategy (even if such individual variation could have evolutionary meaning). With the exception of shorter menstrual cycles found in one study, the traits Rushton cites as r-selected in association with twinning are either statistical artifacts of no reproductive value or figments of misinterpretation.
Conclusion
I have discussed a few biological variables that lead to higher rates of twinning and I have cited some research which shows that black women have higher rates of some of the hormones that are related to higher rates of twinning. But I have also shown that it’s not so simple to jump to a genetic conclusion, since these hormones are of course mediated by environmental factors like diet.
Rushton quite clearly takes these twinning rate differences to be “genetic” in nature, but we are in the 2020s now, not the 1980s, and we now know that genes are necessary, but passive players in the formation of phenotypes (Noble, 2011, 2012, 2016; Richardson, 2017, 2021; Baverstock, 2021; McKenna, Gawne, and Nijhout, 2022). These new ways of looking at genes—as passive, not active causes, and as not special from any other developmental resources—shows how the reductionist thinking of Rushton and his contemporaries were straight out false. Nonetheless, while Rushton did get it right that there is a racial difference in twinning, the difference, I think, isn’t a genetic difference and I certainly don’t think they it lends credence to his Differential K theory, since Anderson showed that if we were to accept Rushton’s premises, then African would be K and Asians would be r. So while there also are differences in menarche between blacks and whites, this too also seems to be environmentally driven.
Rushton’s twinning thesis was his “best bet” at attempting to show that his r/K theory was “right” about racial differences. But the numerous devestating critiques of not only Rushton’s thesis on twinning but his r/K Differential K theory itself shows that Rushton was merely a motivated reasoner (David Duke also consulted with Rushton when Duke wrote his book My Awakening, where Duke describes how psychologists led to his “racial awakening”), so “The claim that Rushton was acting only as a scientist is not credible given this context” (Winston, 2020). Even the usefulness of psychometric life history theory has been recently questioned (this derives from Rushton’s Differential K, Sear, 2020).
But it is now generally accepted that Rushton’s r/K and the current psychometric life history theory that rose from the ashes of Rushton’s theory just isn’t a good way to conceptualize how humans live in the numerous biomes we live in.
Psychologist J. P. Rushton was perhaps most famous for attempting to formulate a grand theory of racial differences. He tried to argue that, on a matrix of different traits, the “hierarchy” was basically Mongoloids > Caucasoids > Negroids. But Rushton’s theory was met with much force, and many authors in many of the different disciplines in which he derived his data to formulate his theory attacked his r/K selection theory also known as Differential K theory (where all humans are K, but some humans are more K than others, so some humans are more r than others). Nonetheless, although his theory has been falsified for many decades, did he get some things right about race? Well, a stopped clock is right twice a day, so it wouldn’t be that outlandish to believe that Rushton got some things right about racial differences, especially when it comes to physical differences. While we can be certain that there are physical differences in groups we term “racial groups” and designate “white”, “black”, “Asian”, “Native American”, and “Pacific Islander” (the five races in American racetalk), this doesn’t lend credence to Rushton’s r/K theory.
In this article, I will discuss Rushton’s claims on motor development between blacks and whites. I will argue that he basically got this right, but it is of no consequence to the overall truth of his grand theory of racial differences. We know that there are physical differences between racial groups. But that there are physical differences between racial groups doesn’t entail that Rushton’s grand theory is true. The only entailment, I think, that can be drawn from that is there is a possibility that physical differences between races could exist between them, but it is a leap to attribute these differences to Rushton’s r/K theory, since it is a falsified theory on logical, empirical and methodological grounds. So I will argue that while Rushton got this right, a stopped clock is right twice a day but this doesn’t mean that his r/K theory is true for human races.
Was Rushton right? Evaluating newer studies on black-white motor development
Imagine three newborns: one white, one black and the third Asian and you observe the first few weeks of their lives. Upon observing the beginnings of their lives, you begin to notice differences in motor development between them. The black infant is more motorically advanced than the white infant who is more motorically advanced than the Asian infant. The black infant begins to master movement, coordination and dexterity showing a remarkable level of motoric dexterity, while the white infant shows less motoric dexterity than the black infant, and the Asian infant still shows lower motoric dexterity than the white infant.
These disparities in motor development are evidence in the early stages of life, so is it genetic? Cultural? Bio-cultural? I will argue that what explains this is a bio-cultural view, and so it will of course eschew reductionism, but of course as infants grow and navigate through their cultural milieu and family lives, this will have a significant effect on their experiences and along with it their motoric development.
Although Rushton got a lot wrong, it seems that he got this issue right—there does seem to be differences in precocity of motor development between the races, and the references he cites below in his 2000 edition of Race, Evolution, and Behavior—although most are ancient compared to today’s standards—hold to scrutiny today, where blacks walk earlier than whites who walk earlier than Asians.
Revised forms of Bayley’s Scales of Mental and Motor Development administered in 12 metropolitan areas of the United States to 1,409 representative infants aged 1-15 months showed black babies scored consistently above whites on the Motor Scale (Bayley, 1965). This difference was not limited to any one class of behavior, but included: coordination (arm and hand); muscular strength and tonus (holds head steady, balances head when carried, sits alone steadily, and stands alone); and locomotion (turns from side to back, raises self to sitting, makes stepping movements, walks with help, and walks alone).
Similar results have been found for children up to about age 3 elsewhere in the United States, in Jamaica, and in sub-Saharan Africa (Curti, Marshall, Steggerda, & Henderson, 1935; Knobloch & Pasamanik, 1953; Williams & Scott, 1953; Walters, 1967). In a review critical of the literature Warren (1972) nonetheless reported evidence for African motor precocity in 10 out of 12 studies. For example, Geber (1958:186) had examined 308 children in Uganda and reported an “all-round advance of development over European standards which was greater the younger the child.” Freedman (1974, 1979) found similar results in studies of newboms in Nigeria using the Cambridge Neonatal Scales (Brazelton & Freedman, 1971).
Mongoloid children are motorically delayed relative to Caucasoids. In a series of studies carried out on second- through fifth-generation Chinese-Americans in San Francisco, on third- and fourth-generation Japanese-Americans in Hawaii, and on Navajo Amerindians in New Mexico and Arizona, consistent differences were found between these groups and second- to fourth-generation European-Americans using the Cambridge Neonatal Scales (Freedman, 1974, 1979; Freedman & Freedman, 1969). One measure involved pressing the baby’s nose with a cloth, forcing it to breathe with its mouth. Whereas the average Chinese baby fails to exhibit a coordinated “defense reaction,” most Caucasian babies turn away or swipe at the cloth with the hands, a response reported in Western pediatric textbooks as the normal one.
On other measures including “automatic walk,” “head turning,” and “walking alone,” Mongoloid children are more delayed than Caucasoid children. Mongoloid samples, including the Navajo Amerindians, typically do not walk until 13 months, compared to the Caucasian 12 months and Negro 11 months (Freedman, 1979). In a standardization of the Denver Developmental Screening Test in Japan, Ueda (1978) found slower rates of motoric maturation in Japanese as compared with Caucasoid norms derived from the United States, with tests made from birth to 2 months in coordination and head lifting, from 3 to 5 months in muscular strength and rolling over, at 6 to 13 months in locomotion, and at 15 to 20 months in removing garments.
Regarding newer studies on this matter, there are differences between European and Asian children in the direction that Rushton claimed. Infants from Hong Kong displayed a difference sequence of rolling compared to Canadian children. There does seem to be a disparity in motoric development between Asian and white children (Mayson, Harris, and Bachman, 2007). These authors do cite some of the same studies like the DDST (which is currently outdated) which showed how Asian children were motorically delayed compared to white children. And although they put caution on their findings of their literature review, it’s quite clear that this pattern exists and it is a bio-cultural one. So they conclude their literature review writing “the literature reviewed suggests differences in rate of motor development among children of various ethnic origins, including those of Asian and European descent” and that “Limited support suggests also that certain developmental milestones, such as rolling, may differ between infants of Asian and European origin.” Further, cultural practices in northern China—for example, lying them on their backs on sandbags—stall the onset of walking in babies sitting, crawling, and walking by a few months (Karasik et al, 2011).
This is related to the muscles that are used to roll from a supine to prone position and vice versa. Since some Asian children spend a longer time in apparatuses that aren’t conducive to growing a strong muscular base to be able to roll from the supine to prone position, to crawl and eventually walk, this is the “cultural” in the “bio-cultural” approach I will argue for.
One study on Norwegian children found that half of the children were waking by 13 months (the median) while 25 percent were walking by 12 months and 75 percent were walking by 14 months (Storvold, Aarethun, and Bratberg, 2013). One reason for the delayed response time could be supine sleeping, which was put into effect during the Back to Sleep program to mitigate causes of death from SIDS. Although it obviously saved tens of thousands of infant lives, it came at a cost of slightly stunted motoric development. It also seems that there is poor predictive value for infant milestones such as walking when it comes to health (Jenni et al, 2012).
Black Caribbean, black African and Indian infants were less likely to show delays in gross motor milestones compared to white infants. But Pakistani and Bangladeshi infants were more likely to be delayed in motoric development and communicative gestures, which was partly attributed to socio-cultural factors (Kelly et al, 2006). Kelly et al (2006: 828) also warn against genetic conclusions based on their large findings of difference between white and African and Caribbean infants:
The differences we observed between Black African and Black Caribbean compared with White infants are large and remain unaffected after adjusting for important covariates. This makes it tempting to conclude that the remaining effect must be a consequence of genetic differences. However, such a conclusion would be prematurely drawn. First, we have not included the measurement of genetic factors in our analysis, and, therefore, the presence of such effects cannot be demonstrated. Second, speculating on such effects should only be done alongside recognition that the model we have been able to test contains imperfect measurement.
It has also been observed that black and white children achieved greater mastery of motoric ability (locomotor skills) compared to Asian children but there was no difference by age group (Adeyemi-Walker et al, 2018). It was also found that infants with higher motor development scores had a lower weight weight relative to their length as they grew. So it was found that delayed motor development was associated with higher weight relative to length (Shoaibi et al, 2018). Black infants are also more motorically advanced and this is seen at up to two years of age (Malina, 1988) while black children perform better on tests of motor ability than white children (Okano et al, 2001). Kilbride et al (1970) also found that Baganda infants in Uganda showed better motoric ability than white American children. Campbell and Heddeker (2001) also showed that black infants were more motorically advanced than infants of other races.
It is clear that research like this blows up the claim that there should be a “one-size fits all” chart for motoric development in infants and that there should be race-specific milestones. This means that we should throw out the WEIRD assumptions when it comes to motoric development of infants (Karasik et al, 2011). They discuss research in other cultures where African, Caribbean and Indian caregivers massage the muscles of babies, stretch their limbs, toss them in their air, sit them up, and walk with them while helping them which then shapes their muscles and has them learn the mind-muscle connections needed to be able to learn how to eventually walk. And it also seems that random assignment to exercise excelerates how quickly an infant walks. White infants also sit at 6 months while black infants sit at 4 months. Nonetheless, it is clear that culture and context can indeed shape motoric development in groups around the world.
A bio-cultural view of motor development
When it comes to biological influences on motor development, sex and age are two important variables (Escolano-Perez, Sanchez-Lopez, and Herrero-Nivela, 2021). Important to this, of course, is that the individual must be normal, and they must have a normal brain with normal vision and spatial skills. They must be able to hear (to eventually follow commands and hear what is going on in their environment to change their course of action if need be). Further, the child’s home environment and gestational age influence different portions of motoral development (Darcy, 2022). After infants begin crawling, their whole world changes and they process visual motion better and faster, being able to differentiate between different speeds and directions, so a stimulating environment for the infant can spur the development of the brain (Van der Meer and Van der Weel, 2022). Biological maturation and body weight also affect motor development. Walking develops naturally, but walking and motor competence need to be nurtured for the child to reach their full potential; lower motor competence is related to higher body weight (Drenowatz and Greier, 2019).
One study on Dutch and Israeli infants even found—using developmental niche construction—that “infant motor development indeed is at least partly culturally constructed [which] emphasizes the importance of placing infant motor development studies into their ‘cultural cradle‘ (Oudgeneong, Atun-Eni, and Schaik, 2020). Gross motor development—rolling over, crawling, alternating kicks, moving from lying to sitting, and having tummy time—is recognized by the WHO. Further, children from different cultures have different experiences, which also could lead to, for example, not doing things that are conducive to the development of gross motor development (Angulo-Barroso et al, 2010). Moreover, motor development is embodied, enculturated, embedded, and enabling (Adolph and Hoch, 2020). It is also known that differences in the cultural environment “have a non-negligible effect on motor development” (Bril, 1986). Motor development also takes place in physical environments and is purposive and goal-directed (Hallemans, Verbeque, and de Walle, 2020).
So putting this all together, we have conceptualized motor development as a dynamic process which is influenced by a complex interplay of biological and cultural factors (Barnes, Zieff, and Anderson, 1999). Biological factors like sex, age, health, sensory abilities, and socio-cultural factors like home environment and developmental niches explain motor development and differences in them between individuals. The cultural differences, though, can impede motoral development, and not allow one to reach milestones they would have otherwise reached in a different cultural environment, just like if one couldn’t hear or see would have trouble reaching developmental milestones.
Children of course grow up in cultural environments and contexts and so they are culturally situated. So what this means is that both the cultural and social environment the child finds themselves in will of course then influence their physical and mental development and lead them to their milestones they hit which is dictated by the normal biology they have which then is allowed by the socio-cultural environment they are born into. So we have the bio-cultural view on motor development, and beyond the cultural environment the child finds themselves in, the interactions they have between parents and caregivers—more knowledgeable others—can be pertinent to their motor development and reaching of developmental milestones. Cultural practices and expectations could emphasize certain milestones over others and then guide the child towards the trajectory. So the framework recognizes that normal biology and sensory perceptions are needed for the development of normal motor development, but that cultural and social differences in that context will spur motor development in the child who finds themselves in different cultures.
Conclusion
Was Rushton right about this? Yes, I think he was. The recent literature on the matter speaks to this. But that doesn’t mean that his r/K selection theory is true. There are differences in motor development between races. But what is interesting is the interaction between biological and cultural factors that spur motor development. The question of black motor precocity, however, is a socio-political question, since science is a social convention influenced by the values of the scientist in question. Now, to the best of my knowledge, Rushton himself never carried out studies on this, he just collated them to use them for his racial trait matrix. However, it’s quite clear that Rushton was politically politically and socially motivated to prove that his theory was true.
So obviously, what is considered “normal” is different in different cultures, and motor development is no different. So just like I think we should have different BMI and skin fold charts for different races, so too should we have different developmental milestones for different races and cultures. The discussion here is clear, since what is “average” and “normal” is different based on race and culture. Like for instance, black babies begin walking around 11 months, white babies around 12 months and Native American babies at 13 months. So while parents may be worried that their child didn’t hit a certain developmental milestone like walking, sitting, rolling, taking a bio-cultural approach will assuage these worries.
Nonetheless, while Rushton was right about race and motor development, we need to center his research project in context. He was clearly motivated, despite the numerous and forceful critiques of his framework, to prove that he was right. But the continuance of Rushton pushing his theory up until his death shows me that he was quite obviously socially and politically motivated, contrary to what he may have said.
We have approached this paper from the stance that science is a social activity, with all observations influenced by, as well as reflective of, the values of scientists and the political leanings of the sociocultural context within which research is conducted. We suggest that when questions of group difference are pursued in science, awareness of how the categories themselves have been shaped by social and historical forces, as well as of the potential effects on society, is important. (Barnes, Zieff, and Anderson, 1999)
Rene Descartes proposed that the peneal gland was the point of contact—the interface—between the immaterial mind and physical body. He thought that the peneal gland in humans was different and special to that of nonhuman animals, where in humans the peneal gland was the seat of the soul (Finger, 1995). This view was eventually shown to be false. However, claims that the mental can causally interact with the physical (interactionist dualism) have been met with similar criticism. If the mental is irreducible to the physical and if the mental does in fact causally interact with the physical, then the mental must be identical with the physical; that is, the mental is reducible to the physical due to physical laws like conservation of energy. This seems to be an issue for the truth of an interactionist dualist theory. But there are solutions. Deny that causal closure of the physical (CCP) is true (the world isn’t causally closed), or argue that CCP is compatible with interactionist dualism, or argue that CCP is question-begging (assuming in a premise what it seeks to establish and conclude) and assumes without proper justification that all physical events must be due to physical causes, which thereby illogically excludes the possibility of mental causation.
In this article I will provide some reasons to believe that CCP is question-begging, and I will argue that mental causation is invisible (see Lowe, 2008). I will also argue that action potentials are the interface by which the mental and the physical interact and which would then lead a conscious decision to make a movement be possible. I will provide arguments that show that interactionist dualism is consistent with physics, while showing that action potentials are the interface that Descartes was looking for. Ultimately, I will show how the mental interacts with the physical for mental causation to be carried out and how this isn’t an issue for the CCP. The view I will argue for here I will call “cognitive interface dualism” since it centers on the influence of mental states on action potentials and on the physical realm, and it conveys the idea that mental processes interface with physical processes through the conduit of action potentials, without implying a reduction of the mental to the physical, making it a substance dualist position since it still adheres to the mental and the physical as two different substances.
Causal closure of the physical
It is claimed that the world is causally closed—this means that every event or occurrence is due to physical causes, all physical events must be due to physical causes. Basically, no non-physical (mental) factors can cause or influence physical events. Here’s the argument:
(1) Every event in the world has a cause. (2) Causes and effects within the physical world are governed by the laws of physics. (3) Non-physical factors or entities, by definition, don’t belong to the physical realm. (4) If a nonphysical factor were to influence a physical event, it would violate the laws of physics. (5) Thus, the world is causally closed, meaning that all causes and effects in it are governed by physical interactions and laws.
But the issue here for the physicalist who wants to use causal closure is the fact that mental events and states are qualitatively different from physical events and states. This is evidenced in Lowe’s distinction between intentional (mental) and event (physical) causation. Mental states like thoughts and consciousness possess qualitatively different properties than physical states. The causal closure argument assumes that physical events are the only causes of other physical events. But mental states appear to exert causal influence over physical events, for instance voluntary action based on conscious decision, like my action right now to write this article. So if M states do influence P events, then there must be interaction between the mental and physical realms. This interaction contradicts the idea of strict causal closure of the physical realm. Since mental causation is necessary to explain aspects of human action and consciousness, it then follows that the physical world may not be causally closed.
The problem of interaction for interactionist dualism is premised on the CCP. It supposedly violated the conservation of energy (CoE). If P energy is needed to do P work, then a convergence of mental into physical energy then results in an increase in energy that is inexplicable. I think there are many ways to attack this supposed knock-down argument against interactionist dualism, and I will make the case in an argument below, arguing that action potentials are where the brain and the mind interact to carry out intentions. However, there are no strong, non-question begging arguments for causal closure that don’t beg the question (eg see Bishop, 2005; Dimitrijevic, 2010; Gabbani, 2013; Gibb, 2015), and the inductive arguments commit a sampling error or non-sequiturs (Buhler, 2020). So the CCP is either question-begging or unsound (Menzies, 2015). I will discuss this issue before concluding this article, and I will argue that my argument that APs serve as the interface between the mental and the physical, along with the question-beggingness of causal closure actually strengthens my argument.
The argument for action potentials as the interface between the mind and the brain
The view that I will argue for here, I think, is unique and has never been argued for in the philosophical literature on mental causation. In the argument that follows, I will show how arguing that action potentials (APs) are the point of contact—the interface—between the mind and brain doesn’t violate the CCP nor does it violate CoE.
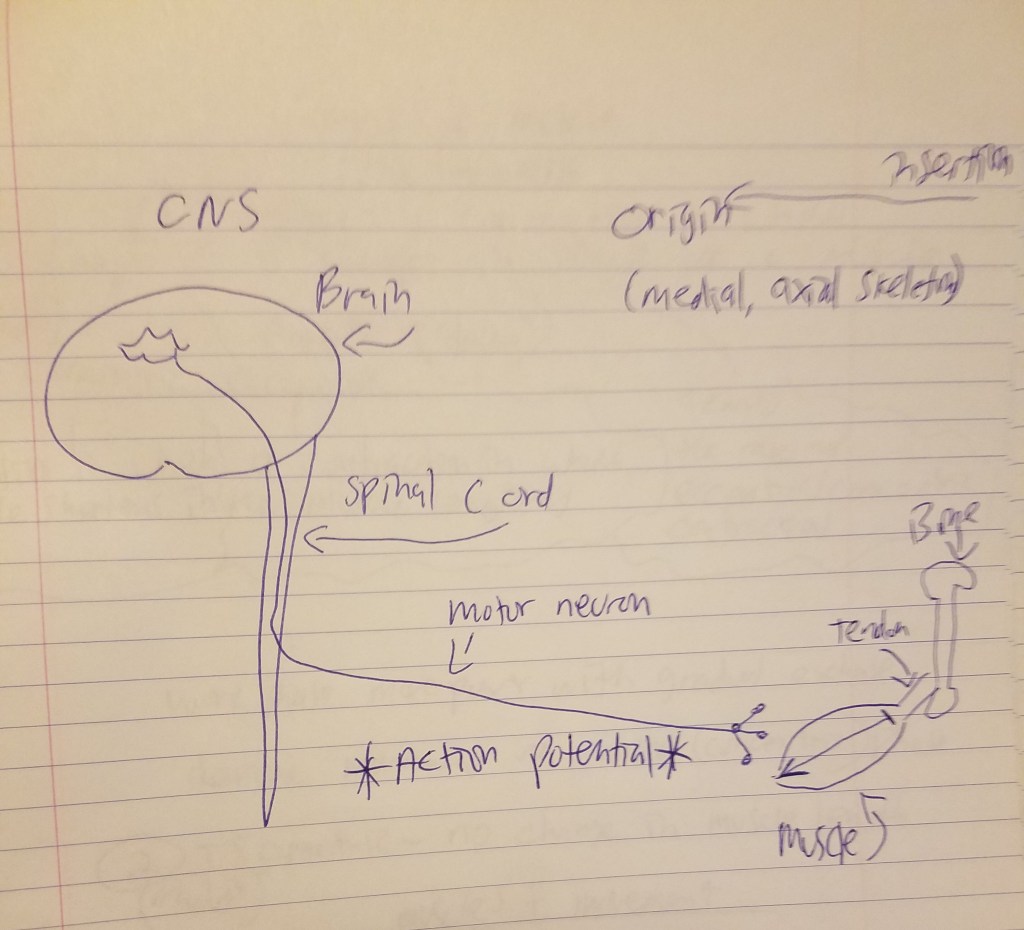
But the skeletal muscle will not contract unless the skeletal muscles are stimulated. The nervous system and the muscular system communicate, which is called neural activiation—defined as the contraction of muscle generated by neural stimulation. We have what are called “motor neurons”—neurons located in the CNS (central nervous system) which can send impulses to muscles to move them. This is done through a special synapse called the neuromuscular junction. A motor neuron that connects with muscle fibers is called a motor unit and the point where the muscle fiber and motor unit meet is callled the neuromuscular junction. It is a small gap between the nerve and muscle fiber called a synapse. Action potentials (electrical impulses) are sent down the axon of the motor neuron from the CNS and when the action potential reaches the end of the axon, hormones called neurotransmitters are then released. Neurotransmitters transport the electrical signal from the nerve to the muscle.
So action potentials (APs) are carried out at the junction between synapses. So, regarding acetylcholine, when it is released, it binds to the synapses (a small space which separates the muscle from the nerve) and it then binds onto the receptors of the muscle fibers. Now we know that, in order for a muscle to contract, the brain sends the chemical message (acetylcholine) across synapses which then initiates movement. So, as can be seen from the diagram above, the MMC refers to the chemo-electric connection between the motor cortex, the cortico-spinal column, peripheral nerves and the neuromuscular junction. A neuromuscular junction is a synapse formed by the contact between a motor neuron and a muscle fiber.
This explanation will set the basis for my argument on how action potentials are the interface—the point of contact—by which the mind and brain meet.
As I have already shown, APs are electrochemical events that transmit signals within the nervous system and are generated as the result of neural activity which can be influenced by mental states like thoughts and intentions. The brain operates in accordance with physical laws and obeys the CoE, the initiation of APs could be (and are, though not always) influenced by mental intentions and processes. Mental processes could modulate the threshold or likelihood of AP firing through complex biomechanical mechanisms that do not violate the CoE. Of course, the energy that is required for generating APs ultimately derives from metabolic processes within the body, which could be influenced by mental states like attention, intention and emotional states. This interaction between mental states does not violate the CoE, nor does it require a violation of the laws of physics, since it operates within the bounds of biochemical and electrochemical processes that respect the CoE. Therefore, APs serve as the point of controlled interaction between the mental and physical realms, allowing for mental causation without disrupting the overall energy balance in the physical world.
Lowe argued that mental causation is invisible, and so since it is invisible, it is not amenable to scientific investigation. This view can be integrated into my argument that APs serve as the interface between the two substances, mental and physical. APs are observable electrochemical events in a neuron which could be influenced by mental states. So as I argued above, mental processes could influence or modulate the veneration of APs. When it comes to the invisibility of mental causation, this refers to the idea that mental events like thoughts, intentions, and consciousness are not directly perceptible like physical objects or events are. Mental states are not observable in the same way that physical events or objects are. In my view, APs hold a dual role. They function as the interface between the mental and the physical, providing the means by which the mental can influence physical events while shaping APs, and they also act as the causal mechanism in connecting mental states to physical events.
Thus, given the distinction between physical events (like APs) and the subjective nature of mental states, the view I have argued for above is consistent with the invisibility of mental causation. Mental causation involves the idea that mental states can influence physical events, and that they have causal efficacy on the physical world. So our mental experiences can lead to physical changes in the world based on the actions we carry out. But since mental states aren’t observable like physical states are, it’s challenging to show how they could lead to effects on the physical world. We infer the influence of mental states on physical events through the effects on observable physical processes. We can’t directly observe intention, we infer it on the basis of one’s action. Mental states could influence physical events through complex chains of electrochemical and biochemical processes which would then make the causative relationship less apparent. So while APs serve as the interface, this doesn’t mean that mental states and APs are identical. This is because while the mental can’t be reduced to physiology (the physical), it encompasses a range of subjective experiences, emotions, thoughts, and intentions that transcend the mechanistic explanations of neural activity.
It is quite obviously an empirical fact that the mental can influence the physical. Think of the fight-or-flight response. When one sees something that they are fearful of (like, say, an animal), there is then a concurrent change in certain hormones. This simple example shows how the mental can have an effect on the physical—where the physical event of seeing something fearful (which would be also be a subjective experience) would then lead to a physical change. So the initial mental event of seeing something fearful is a subjective experience which occurs in the realm of consciousness and mental states. The subjective experience of fear then triggers the fight-or-flight response, which leads to the release of stress hormones like cortisol and adrenaline. These physiological changes are part of the body’s response to a perceived threat based on the subject’s personal subjective experience. So the release of stress hormones is a physical event, and these hormones then have a measurable effect on the body like an increase in heart rate, heightened alertness and energy mobilization which then prepares the subject for action. These physiological changes then prepare the subject to either fight or flee from the situation that caused them fear. This is a solid example on how the mental can influence the physical.
The only way, I think, that my view can be challenged is by arguing that the CCP is true. But if it is question-begging, then my proposition that mental states can influence APs is then less contentious. Furthermore, my argument on APs could be open to multiple interpretations of causal closure. So instead of strictly adhering to causal closure, my view could accommodate various interpretations that allow mental causation to have an effect in the physical realm. Thus, since I view causal closure as question begging, it provides a basis for my view that mental states can influence APs and by extension the physical world. And if the CCP is false, my view on action potentials is actually strengthened.
The view I have argued for here is a simplified perspective on the relationship between the mental and the physical. But my intention isn’t to offer a comprehensive account of all aspects of mental and physical interaction, rather, it is to highlight the role of APs as a point of connection between the mental and physical realms.
Cognitive interface dualism as a form of substance dualism
The view I have argued for here is a substance dualist position. Although it posits an intermediary in APs that facilitates interaction between the mental and physical realms, it still maintains the fundamental duality between mental and physical substances. Mental states are irreducible to physical states, and they interact though APs without collapsing into a single substance. Mental states involve subjective experiences, intentionality, and qualia which are fundamentally different from the objective and quantifiable nature of the physical realm, which I have argued before. APs serve as the bridge—the interface—between the mental and the physical realms, so my dualistic perspective allows for interaction while still preserving the unique properties of the mental and the physical.
Although APs serve as the bridge between the mental and the physical, the interaction between mental states and APs suggests that mental causation operates independently of physical processes. This, then, implies that the self which originates in mental states, isn’t confined to the physical realm, and that it isn’t reducible to the physical. The self’s subjective experiences, consciousness and self-awareness cannot be explained by physical or material processes, which indicates an immaterial substance beyond the physical. The unity of consciousness, which is the integrated sense of self and personal identity over time, are better accounted for by an immaterial self that transcends a change in physical states. Lastly mental states possess qualitative properties like qualia that defy reduction to physical properties. These qualities then, point to a distinct and immaterial self.
My view posits a form of non-reductive mental causation, where mental states influence APs, acknowledging the nonphysical influence on the mental to the physical. Interaction doesn’t imply reduction; mental states remain irreducible even though they impact physical processes. My view also accommodates consciousness, subjectivity, and intentionality which can’t be accounted for by material or physical processes. My view also addresses the explanatory gap between objective physical processes and subjective mental processes, which can’t be accounted for by reduction to physical brain (neural) processes.
Conclusion
The exploration of APs within the context of cognitive interface dualism offers a perspective on the interplay between the mental and physical substances. My view acknowledges APs as the bridge of interaction between the mental and the physical, and it fosters a deeper understanding of the role of mental causation in helping us understand reality.
Central to my view is recognizing that while APs do serve as the interface or conduit by which the mental and the physical interact, and how mental states can influence physical events, this does not entail that the mental is reducible to the physical. My cognitive interface dualism therefore presents a nuanced approach that navigates the interface between the seen and the unseen, the physical and the mental.
Traditional views of causal closure may raise questions about the feasibility of mental causation, the concept’s rigidity is challenged by the intermediary role of APs. While I do hold that the CCP is question-begging, the view I have argued for here explores an alternative avenue which seemingly transcends that limitation. So even if the strict view of the CCP were to fall, my view would remain strong.
This view is also inherently anti-reductionist, asserting that personal identity, consciousness, subjectivity and intentionality cannot be reduced to the physical. Thus, it doesn’t succumb to the traditional limitations of physicalism. Cognitive interface dualism also challenges the notion that we are reducible to our physical brains or our mental activity. The self—the bearer of mental states—isn’t confined to neural circuitry, although the physical is necessary for our mental lives, it isn’t a sufficient condition (Gabriel, 2018).
Lastly, of course this view means that since the mental is irreducible to the physical, then psychometrics isn’t a measurement enterprise. Any argument that espouses the view that the mental is irreducible to the physical would entail that psychometrics isn’t measurement. So by acknowledging that mental states, consciousness, and subjective experiences transcend the confines of physical quantification, cognitive interface dualism dismantles the assumption that the human mind can be measured and encapsulated using numerical metrics. This view holds that the mental resists quantification, since only the physical is quantifiable since only the physical have specified measured objects, objects of measurement and measurement units.
All in all, my view I title cognitive interface dualism explains how mental causation occurs through action potentials. It still holds that the mental is irreducible to the physical, but that the mental and physical interact without M being reduced to P. This view I have espoused, I think, is unique, and it shows how mental causation does occur, it shows how we perform actions.
Many people believe that a thing called “the G-spot”—Grafenberg spot—exists. The g-spot has been referred to as the female prostate (Puppo, 2014) and it also has been theorized that it is an extension of the clitoris (O’Connell et al, 2005). Recent debates in the urology literature are raging, with one side saying that the g-spot exists while the other side says it does not.
In the 1660s, anatomist and physiologist Regnier de Graaf studied male testicles. In 1668, he made a drawing of dissected male testicles, theorizing that the tubule of the epididymus was necessary for sperm to ejaculate into the vagina, since they knew at the time that the testes was necessary for what was later to be called spermatoza. Now we know that he was right (Turner, 2015). He also correctly theorized that the genesis of life is within the fertilized egg, so he “was the first researcher to solve the mystery of reproduction” (Thiery, 2009). It is possible that de Graaf had knowledge of the erogenous zone inside of the vagina that caused immense sexual pleasure when enough pressure was put on it, but it was the German gynecologist Ernst Grafenberg who identified what would today be known as “the g-spot” in the anterior wall of the vagina (Rabinerson et al, 2007; Edwards, 2022). But recently there have been many papers that attempt to show that it is either reality or a myth. Does it exist?
In 1981, Perry and Whipple taught kegel exercises to women to help their stress urinary incontinence. Women who had lost fluid through their urethra had strong pelvic floor muscles while women who had stress urinary incontinence had weak pelvic floor muscles. Women with strong pelvic floor muscles reported that they lost fluid from their urethra during sexual stimulation and some reported it even during orgasm. So they then found that women who had stronger pelvic floor muscles experience were more likely to experience female ejaculation compared to women with weak pelvic floor muscles. They reported feeling it in the anterior wall of the vagina, and they found that when their anterior wall of their vagina was stimulated with two fingers using a “come here motion”, swelled when stimulated. They then called it the “Grafenberg spot”, or g-spot (Whipple, 2015).
Whether or not the g-spot exists has implications for whether or not there is a distinction between clitoral and vaginal orgasms. If the spot is real, then vaginal orgasms are possible. But if the spot is not real, then vaginal orgasms are impossible and all orgasms are clitoral orgasms. Puppo et al (2015) claim that the so-called vaginal orgasms that women report are “always caused by the surrounding erectile organs (triggers of female orgasm).” What Puppo (2014) calls “the female penis; female erectile organs” “are believed to be responsible for female orgasm.” (also see Whipple, 2015). The g-spot is said to be located below the front of the vagina. Schubach (2002) claims there is identity between the female prostate, g-spot, and Skene’s gland, while also stating that the g-spot is not really a “spot” but more of an area. Using histology, Thabet et al (2009) showed that about 18 percent of their sample of Egyptian women didn’t have a g-spot.
The g-spot is basically said to be a vaginal erogenous zone that, when sufficiently stimulated, can produce a vaginal orgasm independent of clitoral stimulation. But Mollaioli et al (2021) reviewed the history of the vagina in reproductive anatomy, stating that it was once thought that the vagina was an inert organ only for delivering babies. They conclude:
that the G-spot surely exists and is present, developed, and active on a tremendously individual basis. However, it is not a spot, and to reduce the risks of misinterpretations and vacuous discussions, it cannot be called G anymore. It is indeed a functional, hormone-dependent area, which may trigger VAOs and in some cases also FEs, well defined as CUV.
There is also what is termed the “A-spot”, which is the anterior fornix erogenous zone, and is said to be 2 inches above the g-spot, the “U-spot”, which is above the urethral opening, and the “C-spot” (clitourethrovaginal complex) (Jannini et al, 2014; Vieira-Baptista, 2021). While it is generally accepted that the anterior vaginal wall is the most sensitive part of the vagina, there seems to be no clear-cut anatomic thing—despite claims to the contrary—that can be termed a “g-spot” in the vagina. Now, this doesn’t mean that vaginal orgasms aren’t a thing, as many women can attest to.
The g-spot is defined as a physiological response, but it has no apparent anatomic correlate and if there is a physiologic response, then there must be an anatomic correlate that allows the physiologic response according to Ostrzenski (2019). Using a cadaver, Ostrzenski (2012) observed that it does indeed have an anatomic structure, near the upper part of the urethral meanus. He observed that it “appeared as a well-delineated sac“, which has anatomic similarities to erectile tissue. But he has some financial conflicts of interest here, since he, as a gynecologist, runs a “g-spot fat augmentation and g-spot surgical augmentation”, offering a plastic surgery intervention (Herold et al, 2015; Ostrzenski, 2018; Triana, 2019).
Reviews of the “spot” agree that there is no single anatomic area in the vagina that we can call “the g-spot” (Jannini at al, 2014; Vieira-Baptista, 2021). But Maratos et al (2015) showed that there is evidence for an “in vivo morphological correlate” of the g-spot and that it’s visibility in MRI can be enhanced using certain techniques (also see Wylie, 2016). Hoag et al (2017) argue that there is no discrete anatomic entity that can be putatively termed as a “g-spot”, but Ostrzenski (2018) claims that the “spot” is observable in their Hoag et al’s figure 4A. In a study of 309 Turkish women, about half of the sample (n=151) stated that the g-spot does exist, and those that had a belief in it had better scores in genital perception and sexual functioning (Kaya and Caliskan, 2018). Buisson et al (2010) used an ultrasound on a volunteer couple to ascertain the existence of the g-spot. They observed the penis inflating the vagina, which then led to a stretched clitoral root that “has consequently a very close relationship with the anterior vaginal wall. This could explain the pleasurable sensitivity of this anterior vaginal area called the G-spot.” This shows the importance of what one thinks about the g-spot and their sexual satisfaction. However, Sivaslioglu et al (2021) studied live tissue (not tissue from a cadaver) and concluded that there is no g-spot on the anterior vaginal wall.
In an ultrasonographic study, Gravina et al (2008) observed a correlation of .863 between the thickness of the distal urethrovaginal segment and vaginal orgasm and a .884 correlation between vaginal wall thickness and the likelihood of experiencing a vaginal orgasm. They also found that women with a thinner vaginal wall were less likely to report having a vaginal orgasm. This could be explained by more nerve endings in women who have thicker vaginal walls.
It is claimed that women who prefer longer penises are more likely to achieve a vaginal orgasm (Costa, Miller, and Brody, 2012; evo-psycho Geoffrey Miller is also an author on this paper. There is a just-so story there saying that the female orgasm could be an adaptation or byproduct (see Puts, Dawood and Welling, 2012 and Wheatley and Puts, 2015). However, adaptationist hypotheses are nothing more than just-so stories, and this is another example of panglossian thinking. In any case, 95 percent of women report clitoral orgasm, 65 percent of women report vaginal orgasm and 35 percent of women report an orgasm due to stimulation of the cervix (Jannini at al, 2019).
Other researchers reject the claim that there is one spot that causes a vaginal orgasm, and that the vagina is not passive, but is dynamically active in causing pleasure to the woman. Due to the anatomic and dynamic relationships between the clitoris, urethra, and anterior vaginal walls (where the spot is hypothesized to be located), this “led to the concept of a clitourethrovaginal (CUV) complex, defining a variable, multifaceted morphofunctional area that, when properly stimulated during penetration, could induce orgasmic responses” (Jannini et al, 2014).
Conclusion
The debate on the existence of the g-spot and vaginal orgasms continues with no clear-cut answer in the literature. It is such a vexing question, and there are many people with many different views on its structure and physiology. I think that the CUV complex is a better candidate than an actual localized “spot” or “button” in the vagina, as it speaks to the dynamicness of the vagina. Pfaus et al (2016) conclude:
The distinction between different orgasms, then, is not between sensations of the external clitoris and internal vagina, but between levels of what a woman understands a ‘whole’ orgasm to consist of. This depends on the experience with direct stimulation of the external clitoris, internal clitoris, and/or cervix, but also with knowledge of the arousing and erotic cues that predict orgasm, knowledge of her own pattern of movements that lead to it, and experience with stimulation of multiple external and internal genital and extra-genital sites (e.g. lips, nipples, ears, neck, fingers, and toes) that can be associated with it. Orgasms do not have to come from one site, nor from all sites; and they do not have to be the same for every woman, nor for every sexual experience even in the same woman, to be whole and valid. And it is likely that such knowledge changes across the lifespan, as women experience different kinds of orgasms from different types of sensations in different contexts and/or with different partners. Thus, what constitutes a ‘whole’ orgasm depends on how a woman sums the parts and the individual manner in which she scales them along flexible dimensions of arousal, desire, and pleasure. The erotic body map a woman possesses is not etched in stone, but rather is an ongoing process of experience, discovery, and construction which depends on her brain’s ability to create optimality between the habits of what she expects and an openness to new experiences.
The distal part of the anterior vaginal wall appears to be the most sensitive region of the vagina, yet the existence of an anatomical “G-spot” on the anterior wall remains to be demonstrated. Objective investigative measures, either not available or not applied when Hines first published his review article over a decade ago, still fail to provide irrefutable evidence for the G-spot’s existence. This may be, in part, because of the extreme variability of the female genitalia on an individual level or, more likely, that this mythical location does not exist.
I think there is something to the anterior vaginal wall that would lead to full-body orgasms, but Hoch (1986) states that “the entire anterior vaginal wall” was “found to be erotically sensitive in most of the women examined.” It is indeed accepted that the anterior vaginal wall is the most sensitive part of the vagina, but that doesn’t mean that the g-spot is a thing (Pan et al, 2015). Ling et al (2014) showed that the proximal and distal third of the anterior vaginal wall were had more innervations (nerve endings) and better vascularization, which implies that the vagina may have a sex-sensitive function just like the clitoris. Song et al, 2015 also showed that the distal part of the anterior vaginal wall had more innervations in “seven fresh Korean cadavers.” But such studies of vaginal innervation were noted in one review to be contradictory (Vieira-Baptista et al, 2021).
The experiences of women who claim to have had a vaginal orgasm should not be discarded, but it is possible that as a paper cited noted above, that it’s merely a clitoral orgasm too. Nevertheless, I don’t see this debate settled anytime soon, and both sides have good arguments. What I think would be best is to just accept the C-spot, clitourethrovaginal complex, and this is a larger erogenous zone—not a spot—comprised of the urethra, vaginal wall, paraurethral glands, and the root of the clitoris, since most of the clitoral components are under the skin (Pauls, 2015). Though Puppo (2015) claims that the entire clitoris “is an external organ.” However, there seem to be “clitoral bulbs” in between the cura and vaginal wall (O’Connell et al, 2005).
The term ‘racism’ has many definitions. What does it mean for a person to be a ‘racist’? What does it mean for a person to have ‘racist beliefs’? What does the term ‘racism’ refer to? The answers to these questions then will inform the next part—what does racism have to do with stress and physiology?
What is ‘racism’?
Racism has many definitions, so many—and so many for uses in different contexts—that it has been argued, for example by those in the far-right, that it is, therefore, a meaningless term. However, just because there are many definitions of the term, it does not then mean that there is no referent for the term we use. A referent is a thing that is signified. In this instance, what is the referent for racism? I will provide a few on-hand definitions and then discuss them.
Logically, it would seem as though ideas about race would have to precede racism. But the subject of racism is more broad and complicated than the subject of race, for at least these two historical reasons. First, the kind of prejudice (prejudged cognitions and negative emotions) and discrimination (treating people differently on the grounds of group identities) that constitute racism have a longer history than the modern idea of race, for instance in European anti-Semitism. And second, insofar as modern ideas of race have been in the service of the dominant interests in international and internal interactions, these ideas of race are ideologies that have devalued non-white groups. That is, ideas of race are themselves already inherently racist.
In philosophy, racism has been treated as attitudes and actions of individuals that affect nonwhites unjustly and social structures and institutions that advantage whites and disadvantage nonwhites. The first is hearts-and-minds or classic racism, for instance the use of stereotypes and harmful actions by whites against people of color, as well as negative feelings about them. The second is structural racism, for instance the use of stereotypes or institutional racism, for instance, the facts of how American blacks and Hispanics are, compared to whites, worse off on major measures of human well-being, such as education, income, family wealth, health, family stability, longevity, and rates of incarceration.
John Lovchik in his book Racism: Reality Built on a Myth (2018: 12) notes that “racism is a system of ranking human beings for the purpose of gaining and justifying an unequal distribution of political and economic power.” Note that using this definition, “hereditarianism” (the theory that individual differences between groups and individuals can be reduced to genes; I will give conceptual reasons why hereditarianism is false as what I hope is my final word on the debate) is a racist theory as it attempts to justify the current social hierarchy. (The reason why IQ tests were first brought to America and created by Binet and Simon; see The History and Construction of IQ Testsand The Frivolousness of the Hereditarian-Environmentalist IQ Debate: Gould, Binet, and the Utility of IQ Testing.) This is why hereditarianism saw its resurgence with Jensen’s infamous 1969 paper. Indeed, many prominent hereditarians have held racist beliefs, and were even eugenicists espousing eugenic ideas.
Headley (2000) notes a few definitions of racism—motivational, behavioral, and cognitive racism. Motivational racism is “the infliction of unequal consideration, motivated by the desire to dominate, based on race alone“; behavioral racism is “failure to give equal consideration, based on the fact of race alone”; and cognitive racism is “unequal consideration, out of a belief in the inferiority of another race.”
I have presented six definitions of racism—though there are many more. Now, for the purposes of this article, I will present my own: the ‘inferiorization’ of a racialized group which is then used to explain disparities in things like IQ test scores, social class/SES, education differences, personality, etc. Now, knowing what we know about physiological systems and how they react to the environment around them—the immediate environment and the social environment—how does this then relate to stress and physiology?
Racism, stress, and physiology
Now that we know what racism is, having had a rundown of certain definitions of ‘racism’, I will now discuss the physiological effects such stances could have on groups racialized as ‘races’ (note that I am using socialraces in this article; recall that social constructivists about race need to be realists about race).
The term ‘weathering’ refers to the body’s breaking down due to stress over time. Such stressors can come from one’s immediate environment (i.e., pollution, foodstuffs, etc) or their social environment (a demanding job, how one perceives themselves and how people react to them). So as the body experiences more and more stress it becomes more and more ‘weathered’ which then leads to heightened risk for disease in stressed individuals/populations.
Allostatic states “refer to altered and sustained activity levels of the primary mediators (e.g., glucocorticosteroids) that integrate energetic and associated behaviours in response to changing environments and challenges such as social interactions, weather, disease, predators and pollution” (McEwen, 2005). Examples of allostatic overload such as acceleration of atherosclerosis, hypertension (HTN), stroke, and abdominal obesity (McEwen, 2005) are more likely to be found in the group we racialize as ‘black’ in America—particularly women (Gillum, 1987; Gillum and Hyattsville, 1996; Barnes, Alexander, and Staggers, 1997; Worral et al, 2002; Kataoka et al, 2013).
Geronimus et al (2006) set to find out whether or not the heightened rate of stressors (e.g., racism, environmental pollution, etc) can explain why black bodies are more ‘weathered’ than white bodies. They found that such differences were not explained by poverty, indicating that it even affects well-off blacks. Allostatic load refers to heightened hormonal production in response to stressors. We know that physiological is homeodynamic and therefore changes based on the immediate environment and social environment (for example, when you feel like you’re about to get into a fight, your heart rate increases and you get ready to ‘fight or flight’).
Experiencing racism (environmental stimuli; real or imagined, the outcome is the same) is associated with increased blood pressure (HTN). So if one experiences racism they will them experience an increase in blood pressure, as BP is a physiological variable (Armstead et al, 1987; McNeilly et al, 1995; see Doleszar et al, 2018 for a review). The concept of weathering, then, shows that racial health disparities are, in fact, racist health disparities (Sullivan, 2015: 106). Racism, then, contributes to higher levels of allostasis and, along with it, higher levels of certain hormones associated with higher allostasis.
One way to measure biological age is by measuring the length of telomeres. Telomeres are found at the ends of chromosomes. Since telomere lengths shorten with age (Shammas, 2012), those with shorter telomeres are ‘biologically older’ than those of the same age with longer telomeres. Geronimus et al (2011) showed that black women had shorter telomeres than white women, which was due to subjective and objective stressors (i.e., racism). Black women in the age group 49-55 were 7.5 years ‘older’ than white women. Thus, they had an older physiological age compared to their chronological age. It is known that direct contact with discriminatory events is associated with poor health outcomes. Harrell, Hall, and Taliaferro (2003) note that:
“…physiological set points and the mechanisms governing them are not fixed. External stressors can permanently alter physiological functioning. Racism increases the volume of stress one experiences and may contribute directly to the physiological arousal that is a marker of stress-related diseases.
Social factors can, indeed, influence physiology and there is a wealth of information on how the social becomes biological and how environmental (social) factors influence physiological systems. Forrester et al (2019) replicated Geronimus’ findings, showing that blacks have a higher ‘biological age’ than whites and that psychosocial factors affect blacks more than whites. Simons et al (2020) also replicated Geronimus’ findings, showing that persistent exposure to racism was associated with higher rates of inflammation in blacks which then predicted higher rates of disease in blacks compared to whites. Such discrimination can help to explain differences in birth outcomes (e.g., Jasienska, 2009), stress, inflammation, obesity, stroke rates, etc in blacks compared to whites (Molnar, 2015).
But what is the mechanism by which higher allostatic load scores contribute to negative outcomes and shorter telomeres indicating a higher biological age? When one feels that they are being discriminated against, the sympathetic nervous system activates due to chronic stress and along with it HPA dysfunction. What this means is that there is a loss of the anti-inflammatory effects of cortisol—it becomes blunted. This then increases oxidative stress and inflammation. Thus, the inflammatory processes result in cardiovascular disease and immune and metabolic dysfunction. The HPA axis monitors and responds to stress—allostatic load. When stress hormones are released, the adrenal gland is targeted. When it receives a signal from the pituitary gland, it pumps epinephrine and norepinephrine into the body, causing our hearts to beat faster, causing us to breathe more deeply—what is known as ‘fight or flight.’ Cortisol is also released and is known as a stress hormone, but when the stressful event is over, all three hormones return to baseline. Thus, the higher amount of stress hormones in circulation indicates higher levels of allostatic load—higher levels of stress in the individual in question. We know that blacks have higher levels of allostatic load (i.e., stress-related hormones) than whites (Duru et al, 2012). Barr (2014: 71-72) writes:
Imagine, though, that before the allostatic load has a chance to return to its baseline level, another stressor is sensed by the hypothalamus. The allostatic load will once again increase to the plateau level. Should the perception of stressors be ongoing, the allostatic load will not have the chance to ever fully recharge, and the adrenal gland will be producing an ongoing stream of stress response hormones. The body will experience chronic elevation in its allostatic load. […] A person experiencing repeated stressors, without the opportunity for intervals that are relatively stress-free, will experience a chronically elevated allostatic load, with higher than normal levels of circulating stress response hormones.
Conclusion
What these studies show, then, is that race is a cause of health inequalities, but it’s not inherent in biology but due to social factors that influence the physiology of the individual in question. The term ‘racism’ has many referents, and using one of them identifies ‘hereditarianism’ as a racist ideology (it is inherently ideological). These overviews of studies show that racial health inequalities are due, in part, to perceived discrimination (racism) thus they are racist health disparities. We know that physiology is a dynamic system that can respond to what occurs in the immediate environment—even the social environment (Williams, 1992). Thus, what explains part of the health inequalities between races is perceived discrimination—racism—and how it affects the body’s physiological systems (HPA axis, HTN, etc) and telomeres.
Yet we get tremendously increased phenotypic variation … because the form and variation of cells, what they produce, whether to grow, to move, or what kind of cell to become, is under control of a whole dynamic system, not the genes. (Richardson, 2017: 125)
In 1976 Richard Dawkins published his groundbreaking book The Selfish Gene (Dawkins, 1976). In the book, Dawkins argues that selection occurs at the level of the gene—“the main theme of his book is a metaphorical account of competition between genes …” (Midgley, 2010: 45). Others then took note of the new theory and attempted to integrate it into their thinking. But is it as simple as Dawkins makes it out to be? Are we selfish due to the genes we carry? Is the theory testable? Can it be distinguished from other competing theories? Can it be used to justify certain behaviors?
Rushton, selfish genes, nationalism and politics
JP Rushton is a serious scholar, perhaps most well-known for attempting to use r/K selection theory to explain human behavior (Anderson, 1991). perhaps has the most controversial use of Dawkins’ theory. The main axiom of the theory is that an organism is just a gene’s way of ensuring the survival of other genes (Rushton, 1997). Thus, Rushton’s formulated genetic similarity theory posits that those who are more genetically similar—who share more genes—will be more altruistic toward those with more similar genes even if they are not related and will therefore show negative attitudes to less genetically similar individuals. This is the gene’s “way” of propagating themselves through evolutionary time. Richardson (2017: 9-11) tells us of all of the different ways in which genes are invoked to attempt to justify X.
In the beginning of his career, Rushton was a social learning theorist studying altruism, even publishing a book on the matter—Altruism, Socialization and Society (Rushton, 1980). Rushton reviews the sociobiological literature and concludes that altruism is a learned behavior. Though, Rushton seems to have made the shift from a social learning perspective to a genetic determinist perspective in the years between the publication of Altruism, Socialization and Society and 1984 when he published his genetic similarity theory. So, attempting to explain altruism through genes, while not part of Rushton’s original research programme, seems, to me, to be a natural evolution in his thought (however flawed it may be).
Skeptic: How do you evaluate the work of Irena”us Eibl-Eibesfeldt, J.P. Rushton, and Pierre van den Berghe, all of whom have argued that kin selection theory does help explain nationalism and patriotism?
Dawkins: One could invoke a kind “misfiring” of kin selection if you wanted to in such cases. Misfirings are common enough in evolution. For example, when a cuckoo host feeds a baby cuckoo, that is a misfiring of behavior which is naturally selected to be towards the host’s own young. There are plenty of opportunities for misfirings. I could imagine that racist feeling could be a misfiring, not of kin selection but of reproductive isolation mechanisms. At some point in our history there may have been two species of humans who were capable of mating together but who might have produced sterile hybrids (such as mules). If that were true, then there could have been selection in favor of a “horror” of mating with the other species. Now that could misfire in the same sort of way that the cuckoo host’s parental impulse misfires. The rule of thumb for that hypothetical avoiding of miscegenation could be “Avoid mating with anybody of a different color (or appearance) from you.”
I’m happy for people to make speculations along those lines as long as they don’t again jump that is-ought divide and start saying, “therefore racism is a good thing.” I don’t think racism is a good thing. I think it’s a very bad thing. That is my moral position. I don’t see any justification in evolution either for or against racism. The study of evolution is not in the business of providing justifications for anything.
This is similar to his reaction when Bret Weinstein remarked that the Nazi’s “behaviors” during the Holocaust “were completely comprehensible at the level of fitness”—at the level of the gene.” To which Dawkins replied “I think nationalism may be an even greater evil than religion. And I’m not sure that it’s actually helpful to speak of it in Darwinian terms.” This is what I like to call “rampant adaptationism.”
This is important because Rushton (1998) invokes Dawkins’ theory as justification for his genetic similarity theory (GST; Rushton, 1997), attempting to justify ethno-nationalism from a gene’s-eye view. Rushton did what Dawkins warned against: using the theory to justify nationalism/patriotism. Rushton (1998: 486) states that “Genetic Similarity Theory explains why” ethnic nationalism has come back into the picture. Kin selection theory (which, like with selfish gene theory, Rushton invoked) has numerous misunderstandings attached to it, and of course, Rushton, too, was an offender (Park, 2007).
Dawkins (1981), in Selfish genes in race or politics stated that “It is annoying to find this elegant and important theory being dragged down to the ephemeral level of human politics, and parochial British politics at that.” Rushton (2005: 494), responded, stating that “feeling a moral obligation to condemn racism, some evolutionists minimised the theoretical possibility of a biological underpinning to ethnic or national favouritism.“
Testability?
The main premise of Dawkins’ theory is that evolution is gene-centered and that selection occurs at the level of the gene—genes that propagate fitness will be selected for while genes that are less fit are selected against. This “genes’-eye view” of evolution states “that adaptive evolution occurs through differential survival of competing genes, increasing the allele frequency of those alleles whose phenotypic trait effects successfully promote their own propagation, with gene defined as “not just one single physical bit of DNA [but] all replicas of a particular bit of DNA distributed throughout the world.“
Noble (2018) discusses “two fatal difficulties in the selfish gene version of neo-Darwinism“:
The first is that, from a physiological viewpoint, it does’t lead to a testable prediction. The only problem is that the central definition of selfish gene theory is not independent of the only experimental test of the theory, which is whether genes, defined as DNA sequences, are in fact selfish, i.e., whether their frequency in the gene pool increases (18). The second difficulty is that DNA can’t be regarded as a replicator separate from the cell (11, 17). The cell, and specifically its living physiological functionality, is what makes DNA be replicated faithfully, as I will explain later.
Noble (2017: 156) further elaborates in Dance to the Tune of Life: Biological Relativity:
Could this problem be avoided by attaching a meaning to ‘selfish’ as applied to DNA sequences that is independent of meanings in terms of phenotype? For example. we could say that a DNA sequence is ‘selfish’ to the extent which its frequency in subsequent generations is increased. This at least would be an objective definition that could be measured in terms of population genetics. But wait a minute! The whole point of the characterisation of a gene as selfish is precisely that this property leads to its success in reproducing itself. We cannot make the prediction of a theory be the basis of the definition of the central element of the theory. If we do that, the theory is empty from the viewpoint of empirical science.
Dawkins’ theory is, therefore “not a physiologically testable hypothesis” (Noble, 2011). Dawkins’ theory posits that the gene is the unit of selection, whereas the organism is only used to propagate the selfish genes. But “Just as Special Relativity and General Relativity can be succintly phrased by saying that there is no global (privileged) frame of reference, Biological Relativity can be phrased as saying that there is no global frame of causality in organisms” (Noble, 2017: 172). Dawkins’ theory privileges the gene as the unit of selection, when there is no direct unit of selection in multi-level biological systems (Noble, 2012).
In The Solitary Self: Darwin and the Selfish Gene, Midgley (2010) states “The choice of the word “selfish” is actually quite a strange one. This word is not really a suitable one for what Dawkins wanted to say about genetics because genes do not act alone.” As Dawkins later noted, “the cooperative gene” would have been a better description, while The Immortal Gene would have been a better title for the book. Midgley (2010: 16) states that Dawkins and Wilson (in The Selfish Gene and Sociobiology, respectively) “use a very simple concept of selfishness derived not from Darwin but from a wider background of Hobbesian social atomism, and give it a general explanation of all behaviour, including that of humans.” Dawkins and others claim that “the thing actually being selected was the genes” (Midgley, 2010: 47).
Dawkins’ theory was repurposed, used to attempt to argue for ethno-nationalism and patriotism—even though Dawkins himself is against such uses. Of course, theories can be repurposed from their original uses, though the use of the theory is itself erroneous, as is the case with regard to Rushton, Russel and Wells (1984) and Rushton (1997, 1998). Since the theory is itself not testable (Noble, 2011, 2017), it should therefore—along with all other theories that use it as its basis—be dropped. While Rushton’s change from social learning to genetic causation regarding altruism is not out of character for his former research (he began his career as a social learning theorist studying altruism; Rushton, 1980), his use of the theory to attempt to explain why individuals and groups prefer those more similar to themselves ultimately fails since it is “logically flawed” (Mealey, 1984: 571).
Genes ‘do’ what the physiological system ‘tells’ them to do; they are just inert, passive templates. What is active is the cell—the genome is an organ of the cell and is what is ‘immortal.’ Genes don’t “control” anything; they are used by and for the physiological system to carry out certain processes (Noble, 2017; Richardson, 2017: chapter 4, 5). There are new views of what ‘genes’ really are (Portin and Wilkins, 2017), what they are and were—are—used for.
Development is dynamic and not determined by genes. Genes (DNA sequences) are followers, not leaders. The leader is the physiological system.
Setting: 50kya in the Environment of Evolutionary Adaptedness
Imagine you and your band are being chased by a group of animals 50kya in the Savanna. You look back as the herd is almost at your band, about to rip them to pieces. When, suddenly, there is a bright, blinding flash and the animals stop in their tracks, giving your band enough time to escape.
Your band looks back about 200 feet away to see a man standing there with his bald head standing at the animals, directing a beam of UV rays at the group. The women stand there, lovestruck, as this man just saved the band.
This man—and men who looked like him—were then taken on many expeditions and the same thing happened whenever the band got into trouble out in the grasslands. A group of animals threatens the band? No problem, there is a bald man there, waiting to use his head to blind the back so the band can get away.
Women then started showing more affection to the bald men as they proved they can save the band, and without having to risk harm, at that. So women started mating more with them. Bald men then gained more power in the society, as the women of course continued to pick berries while the men hunted—bald men going with both groups to give protection.
Then, over time, genes the conferred a higher chance of becoming bald became fixated in the group as it conveyed a fitness benefit from which resulted from protecting the group by shining UV rays at a pack of animals to save the group resulting in women finding it attractive and so having more children with them.
Sound ridiculous? Well, searching for claims for bald heads being an adaptation, I discovered that someone said the same exact thing:
A well-polished bald male head was often used by tribes of cavemen to blind predators. As a result every cavemen hunting group of 8 had one bald member, and thus thousands of years later 1 in 8 men experience early on set of baldness. – Taz Boonsborg, London, UK
There are other similar adaptive stories for bald heads, such as a lather surface area to receive more vitamin D:
Loss of hair creates more skin area, which means more vitamin D can be absorbed from sunlight. This would provide a survival benefit for me, which would explain this trait being passed on.
I wonder if it can be linked to the time in evolution when Europeans lived in Central Asia before moving west to Europe. Vitamin D was a scarce necessity. I like to think of my bald head as sun ray receiver. I have noticed that women 30+ are a lot more likely to be attracted to me partially due to my baldness, sometimes very much so 😉
There are, thankfully, some commenters that say it has no adaptive value. When it comes to the existence of any trait, of course, one can construct a plausible evolutionary narrative to explain the survival of the trait into the current day.
A conversation about baldness should include a conversation about bearded-ness—as I have written about before. The story goes that beards are adaptive for men, since “beards may have been valuable as a threat signal during direct male-versus-male competition for dominance and resources“, while also stating for pattern baldness that “The senescence feature of male pattern baldness may be an advertisement of social maturity. Social maturity includes enhanced social status but decreased physical threat, increased approachability, and a propensity to nurture.” (Muscarella and Cunningham, 1996). They also discuss differing sexual selection theories for pattern baldness, such as “for an increase in the visual area for the intimidation display of reddening color during anger.” So to these authors, baldness signifies social status, and if one is bald or balding, they can then show their emotions more—especially if light-skinned. So baldness is a signal of senescence—biological aging.
Kabai (2008) hypothesizes (storytells) that androgenic alopecia—male pattern baldness (MPB)—is an adaptation. Each hair follicle has its own resistance to whether or not it will fall out. So, to Kabai (2008: 1039), MPB “evolved to elevate UV absorbance and thus to provide some protection against prostate cancer.” This is a classic just-so story. I have written a ton about the relationship between testosterone, prostate cancer (PCa), and vitamin D. Blacks have lower levels of vitamin D, and higher levels of prostate cancer. (It should be notes that Setty et al (1970) showed that MPB is four times less likely in blacks compared to whites.) So, in whites, baldness was adaptive in order to acquire more UV rays.
Unfortunately for Kabai (2008), the underlying logic of his hypothesis (that the more bald a head, the higher the vitamin D production) was tested. Bolland et al (2008: 675) “found no evidence to support the hypothesis that the degree of baldness in influences serum 25-OHD levels.” This could be, as the authors note, due to the fact that vitamin D is not produced in the scalp or that vitamin D is produced in the scalp but getting sunburned would modify a man’s behavior to spend less time in the sun and so, the so-called benefits of a bald head for vitamin D production would be limited. Bolland et al (2008) end up concluding that:
there is no accepted evolutionary explanation for the almost universal prevalence of hair loss in older men. We suggest that other hypotheses are required to determine why older men go bald and whether baldness serves any physiological purpose.
Why must baldness “serve a physiological purpose”? I like how the proposal of these hypotheses doesn’t consider the fact that hair just falls out at a certain rate in certain individuals with no evolutionary purpose behind it. Everything must have an adaptive purpose, it seems, and so, one creates these fantastic stories. It’s just like Rudyard Kipling’s stories, actually.
Lastly, Yanez (2004) proposes a cultural hypothesis for MPB. Yanez (2004: 982) states that the cause of MPB is “the detention in the sebum flow moving towards the root of hair.” Those who are more likely to suffer from baldness are those with thin hair who constantly cut their hair short, or hair that is straight or low-density. On the other hand, those with high hair density and thicker hair are less likely to go bald, even if they keep short hair. This, to Yanez (2004), the catalyst of balding is cultural but, of course, is driven by physiological factors (blocking the flow of sebum to the hair follicle).
Many kinds of stories have been crafted in order to explain how and why humans—mostly men—are bald. Though, this just speaks to the problem with adaptationist hypotheses—notice bald heads; bald heads are still around (obviously, since we are observing it); since we notice bald heads because they are still around then there must have been an evolutionary advantage for bald heads; *advantages noted above*; therefore baldness is an adaptation. This reasoning, though, is faulty—it’s a kind of rampant adaptationism, that if a trait exists today then it must, therefore, procure an advantage in an evolutionary context which was then therefore selected-for.
Of course, we can not—and should not—discard the hypothesis that bald men still exist because they could direct UV rays at oncoming predators trying to kill the band, the women seeing it, and it, therefore, becoming an attractive signal that the man can protect the family by directing UV rays at other men and animals. That’s a good hypothesis that’s worth investigating. (Sarcasm.)
What would you think if you heard about a new fortune-telling device that is touted to predict psychological traits like depression, schizophrenia and school achievement? What’s more, it can tell your fortune from the moment of your birth, it is completely reliable and unbiased — and it only costs £100.
This might sound like yet another pop-psychology claim about gimmicks that will change your life, but this one is in fact based on the best science of our times. The fortune teller is DNA. The ability of DNA to understand who we are, and predict who we will become has emerged in the last three years, thanks to the rise of personal genomics. We will see how the DNA revolution has made DNA personal by giving us the power to predict our psychological strengths and weaknesses from birth. This is a game-changer as it has far-reaching implications for psychology, for society and for each and every one of us.
This DNA fortune teller is the culmination of a century of genetic research investigating what makes us who we are. When psychology emerged as a science in the early twentieth century, it focused on environmental causes of behavior. Environmentalism — the view that we are what we learn — dominated psychology for decades. From Freud onwards, the family environment, or nurture, was assumed to be the key factor in determining who we are. (Plomin, 2018: 6, my emphasis)
The main premise of Plomin’s 2018 book Blueprint is that DNA is a fortune teller while personal genomics is a fortune-telling device. The fortune-telling device Plomin most discusses in the book is polygenic scores (PGS). PGSs are gleaned from GWA studies; SNP genotypes are then added up with scores of 0, 1, and 2. Then, the individual gets their PGS for trait T. Plomin’s claim—that DNA is a fortune teller—though, falls since DNA is not a blueprint—which is where the claim that “DNA is a fortune teller” is derived.
It’s funny that Plomin calls the measure “unbiased”, (he is talking about DNA, which is in effect “unbiased”), but PGS are anything BUT unbiased. For example, most GWAS/PGS are derived from European populations. But, for example, there are “biases and inaccuracies of polygenic risk scores (PRS) when predicting disease risk in individuals from populations other than those used in their derivation” (De La Vega and Bustamante, 2018). (PRSs are derived from statistical gene associations using GWAS; Janssens and Joyner, 2019.) Europeans make up more than 80 percent of GWAS studies. This is why, due to the large amount of GWASs on European populations, that “prediction accuracy [is] reduced by approximately 2- to 5-fold in East Asian and African American populations, respectively” (Martin et al, 2018). See for example Figure 1 from Martin et al (2018):
With the huge number of GWAS studies done on European populations, these scores cannot be used on non-European populations for ‘prediction’—even disregarding the other problems with PGS/GWAS.
By studying genetically informative cases like twins and adoptees, behavioural geneticists discovered some of the biggest findings in psychology because, for the first time, nature and nurture could be disentangled.
[…]
… DNA differences inherited from our parents at the moment of conception are the consistent, lifelong source of psychological individuality, the blueprint that makes us who we are. A blueprint is a plan. … A blueprint isn’t all that matters but it matters more than everything else put together in terms of the stable psychological traits that make us who we are. (Plomin, 2018: 6-8, my emphasis)
These big findings were based on twin and adoption studies that indirectly assessed genetic impact. Twenty years ago the DNA revolution began with the sequencing of the human genome, which identified each of the 3 billion steps in the double helix of DNA. We are the same as every other human being for more than 99 percent of these DNA steps, which is the blueprint for human nature. The less than 1 per cent of difference of these DNA steps that differ between us is what makes us who we are as individuals — our mental illnesses, our personalities and our mental abilities. These inherited DNA differences are the blueprint for our individuality …
[DNA predictors] are unique in psychology because they do not change during our lives. This means that they can foretell our futures from our birth.
[…]
The applications and implications of DNA predictors will be controversial. Although we will examine some of these concerns, I am unabashedly a cheerleader for these changes. (Plomin, 2018: 8-10, my emphasis)
This quote further shows Plomin’s “blueprint” for the rest of his book—DNA can “foretell our futures from our birth”—and how it affects his conclusions gleaned from his work that he mostly discusses in his book. Yes, all scientists are biased (as Stephen Jay Gould noted), but Plomin outright claimed to be an unabashed cheerleader for his work. Plomin’s self-admission for being an “unabashed cheerleader”, though, does explain some of the conclusions he makes in Blueprint.
However, the problem with the mantra ‘nature and nurture’ is that it runs the risk of sliding back into the mistaken view that the effects of genes and environment cannot be disentangled.
The problem with the mantra “nature and nurture” is not that it “runs the risk of sliding back into the mistaken view that the effects of genes and environment cannot be disentangled”—though that is one problem. The problem is how Plomin assumes how DNA works. That DNA can be disentangled from the environment presumes that DNA is environment-independent. But as Moore shows in his book The Dependent Gene—and as Schneider (2007) shows—“the very concept of a gene requires the environment“. Moore notes that “The common belief that genes contain context-independent “information”—and so are analogous to “blueprints” or “recipes”—is simply false” (quoted in Schneider, 2007). Moore showed in The Dependent Gene that twin studies are flawed, as have numerous other authors.
Lewkowicz (2012) argues that “genes are embedded within organisms which, in turn, are embedded in external environments. As a result, even though genes are a critical part of developmental systems, they are only one part of such systems where interactions occur at all levels of organization during both ontogeny and phylogeny.” Plomin—although he does not explicitly state it—is a genetic reductionist. This type of thinking can be traced back, most popularly, to Richard Dawkins’ 1976 book The Selfish Gene. The genetic reductionists can, and do, make the claim that organisms can be reduced to their genes, while developmental systems theorists claim that holism, and not reductionism, better explains organismal development.
The main thrust of Plomin’s Blueprint rests on (1) GWA studies and (2) PGSs/PRSs derived from the GWA studies. Ken Richardson (2017b) has shown that “some cryptic but functionally irrelevant genetic stratification in human populations, which, quite likely, will covary with social stratification or social class.” Richardson’s (2017b) argument is simple: Societies are genetically stratified; social stratification maintains genetic stratification; social stratification creates—and maintains—cognitive differentiation; “cognitive” tests reflect prior social stratification. This “cryptic but functionally irrelevant genetic stratification in human populations” is what GWA studies pick up. Richardson and Jones (2019) extend the argument and argue that spurious correlations can arise from genetic population structure that GWA studies cannot account for—even though GWA study authors claim that this population stratification is accounted for, social class is defined solely on the basis of SES (socioeconomic status) and therefore, does not capture all of what “social class” itself captures (Richardson, 2002: 298-299).
Heritability statistics do remain useful in some limited circumstances, including selective breeding programs in which developmental environments can be strictly controlled. But in environments that are not controlled, these statistics do not tell us much. In light of this, numerous theorists have concluded that ‘the term “heritability,” which carries a strong conviction or connotation of something “[in]heritable” in the everyday sense, is no longer suitable for use in human genetics, and its use should be discontinued.’ 31 Reviewing the evidence, we come to the same conclusion.
Heritability estimates assume that nature (genes) can be separated from nurture (environment), but “the very concept of a gene requires the environment” (Schneider, 2007) so it seems that attempting to partition genetic and environmental causation of any trait T is a fool’s—reductionist—errand. If the concept of gene depends on and requires the environment, then how does it make any sense to attempt to partition one from the other if they need each other?
Let’s face it: Plomin, in this book Blueprint is speaking like a biological reductionist, though he may deny the claim. The claims from those who push PRS and how it can be used for precision medicine are unfounded, as there are numerous problems with the concept. Precision medicine and personalized medicine are similar concepts, though Joyner and Paneth (2015) are skeptical of its use and have seven questions for personalized medicine. Furthermore, Joyner, Boros and Fink (2018) argue that “redundant and degenerate mechanisms operating at the physiological level limit both the general utility of this assumption and the specific utility of the precision medicine narrative.” Joyner (2015: 5) also argues that “Neo-Darwinism has failed clinical medicine. By adopting a broader perspective, systems biology does not have to.”
Janssens and Joyner (2019) write that “Most [SNP] hits have no demonstrated mechanistic linkage to the biological property of interest.” Researchers can show correlations between disease phenotypes and genes, but they cannot show causation—which would be mechanistic relations between the proposed genes and the disease phenotype. Though, as Kampourakis (2017: 19), genes do not cause diseases on their own, they only contribute to its variation.
GPS are unique predictors in the behavioural sciences. They are an exception to the rule that correlations do not imply causation in the sense that there can be no backward causation when GPS are correlated with traits. That is, nothing in our brains, behaviour or environment changes inherited differences in DNA sequence. A related advantage of GPS as predictors is that they are exceptionally stable throughout the life span because they index inherited differences in DNA sequence. Although mutations can accrue in the cells used to obtain DNA, like any cells in the body these mutations would not be expected to change systematically the thousands of inherited SNPs that contribute to a GPS.
Turkheimer goes on to say that this (false) assumption by Plomin and Stumm (2018) assumes that there is no top-down causation—i.e., that phenotypes don’t cause genes, or there is no causation from the top to the bottom. (See the special issue of Interface Focusfor a slew of articles on top-down causation.) Downward causation exists in biological systems (Noble, 2012, 2017), as does top-down. The very claim that “nothing in our brains, behaviour or environment changes inherited differences in DNA sequence” is ridiculous! This is something that, of course, Plomin did not discuss in Blueprint. But in a book that, supposedly, shows “how DNA makes us who we are”, why not discuss epigenetics? Plomin is confused, because DNA methylation impacts behavior and behavior impacts DNA methylation (Lerner and Overton, 2017: 114). Lerner and Overtone (2017: 145) write that:
… it should no longer be possible for any scientist to undertake the procedure of splitting of nature and nurture and, through reductionist procedures, come to conclusions that the one or the other plays a more important role in behavior and development.
Plomin’s reductionist takes, therefore again, fail. Plomin’s “reluctance” to discuss “tangential topics” to “inherited DNA differences” included epigenetics (Plomin, 2018: 12). But it seems that his “reluctance” to discuss epigenetics was a downfall in his book as epigenetic mechanisms can and do make a difference to “inherited DNA differences” (see for example, Baedke, 2018, Above the Gene, Beyond Biology: Toward a Philosophy of Epigenetics and Meloni, 2019, Impressionable Biologies: From the Archaeology of Plasticity to the Sociology of Epigenetics see also Meloni, 2018). The genome can and does “react” to what occurs to the organism in the environment, so it is false that “nothing in our brains, behaviour or environment changes inherited differences in DNA sequence” (Plomin and Stumm, 2018), since our behavior and actions can and do methylate our DNA (Meloni, 2014) which falsifies Plomin’s claim and which is why he should have discussed epigenetics in Blueprint. End Edit
Conclusion
So the main premise of Plomin’s Blueprint is his two claims: (1) that DNA is a fortune teller and (2) that personal genomics is a fortune-telling device. He draws these big claims from PGS/PRS studies. However, over 80 percent of GWA studies have been done on European populations. And, knowing that we cannot use these datasets on other, non-European datasets, greatly hampers the uses of PGS/PRS in other populations—although the PGS/PRS are not that useful in and of itself for European populations. Plomin’s whole book is a reductionist screed—“Sure, other factors matter, but DNA matters more” is one of his main claims. Though, a priori, since there is no privileged level of causation, one cannot privilege DNA over any other developmental variables (Noble, 2012). To understand disease, we must understand the whole system and how when one part of the system becomes dysfunctional how it affects other parts of the system and how it runs. The PGS/PRS hunts are reductionist in nature, and the only answer to these reductionist paradigms are new paradigms from systems biology—one of holism.
Plomin’s assertions in his book are gleaned from highly confounded GWA studies. Plomin also assumes that we can disentangle nature and nurture—like all reductionists. Nature and nurture interact—without genes, there would be an environment, but without an environment, there would be no genes as gene expression is predicated on the environment and what occurs in it. So Plomin’s reductionist claim that “Our future is DNA” is false—our future is studying the interactive developmental system, not reducing it to a sum of its parts. Holistic biology—systems biology—beats reductionist biology—the Neo-Darwinian Modern Synthesis.
DNA is not a blueprint nor is it a fortune teller and personal genomics is not a fortune-telling device. The claim that DNA is a blueprint/fortune teller and personal genomics is a fortune-telling device come from Plomin and are derived from highly flawed GWA studies and, further, PGS/PRS. Therefore Plomin’s claim that DNA is a blueprint/fortune teller and personal genomics is a fortune-telling device are false.
The association between muscle fiber typing obesity and race is striking. It is well-established that blacks have a higher proportion of type II skeletal muscle fibers than whites and these higher proportions of these specific types of muscle fibers lead to physiological differences between the two races which then lead to differing health outcomes between them—along with differences in athletic competition. Racial differences in health are no doubt complex, but there are certain differences between the races that we can look at and say that there is a relationship here that warrants further scrutiny.
Why is there an association between negative health outcomes and muscle phsyiology? The answer is very simple if one knows the basics of muscle physiology and how and why muscles contract (it is worth noting that out of a slew of anatomic and phsyiologic factors, movement is the only thing we can consciously control, compare to menstration and other similar physiologic processes which are beyond our control). In this article, I will describe what muscles do, how they are controlled, muscle physiology, the differences in fiber typing between the races and what it means for health outcomes between them.
Muscle anatomy and physiology
Muscle fiber number is determined by the second trimester. Bell (1980) noted that skeletal muscle fiber in 6 year olds is not different from normal adult tissue, and so, we can say that between the time in the womb and age 6, muscle fiber type is set and cannot be changed (though training can change how certain fibers respond, see below).
Muscle anatomy and physiology is interesting because it shows us how and why we move the way we do. Tendons attach muscle to bone. Attached to the tendon is the muscle belly. The muscle belly is made up of facsicles and the fascicles are made up of muscle fibers. Muscle fibers are made up of myofibrils and myofibrils are made up of myofilaments. Finally, myofilaments are made up of proteins—specifically actin and myosin, this is what makes up our muscles.
Muscle fibers are encased by sarcolemma which contains cell components such as sarcoplasm, nuclei, and mitochondria. They also have other cells called myofibrils which contain myofilaments which are then made up of actin (thin filaments) and mysoin (thick filaments). These two types of filaments form numerous repeating sections within a myofibril and each repeating section is known as a sarcomere. Sarcomeres are the “functional” unit of the muscle, like the neuron is for the nervous system. Each ‘z-line’ denotes another sarcomere across a myofibril (Franzini-Armstrong, 1973; Luther, 2009).
Other than actin and myosin, there are two more proteins important for muscle contraction: tropomyosin and troponin. Tropomyosin is found on the actin filament and it blocks myosin binding sites which are located on the actin filament, and so it keeps myosin from attaching to muscle while it is in a relaxed state. On the other hand, troponin is also located on the actin filament but troponin’s job is to provide binding sites for calcium and tropomyosin when a muscle needs to contract.
So the structure of skeletal muscle can be broken down like so: epymyseum > muscle belly > perimyseum > fascicle > endomyseum > muscle fibers > myofibrils > myofilaments > myosin and actin. Note diagram (C) from above; the sarcomere is the smallest contractile unit in the myofibril. According to sliding filament theory (see Cook, 2004 for a review), a sarcomere shortens as a result of the ‘z-lines’ moving closer together. The reason these ‘z-lines’ converge is because myosin heads attach to the actin filament which asynchronistically pulls the actin filament across the myosin, which then results in the shortening of the muscle fiber. Sarcomeres are the basic unit controlling changes in muscle length, so the faster or slower they fire depends on the majority type of fiber in that specific area.
But the skeletal muscle will not contract unless the skeletal muscles are stimulated. The nervous system and the muscular system communicate, which is called neural activiation—defined as the contraction of muscle generated by neural stimulation. We have what are called “motor neurons”—neurons located in the CNS (central nervous system) which can send impulses to muscles to move them. This is done through a special synapse called the neuromuscular junction. A motor neuron that connects with muscle fibers is called a motor unit and the point where the muscle fiber and motor unit meet is callled the neuromuscular junction. It is a small gap between the nerve and muscle fiber called a synapse. Action potentials (electrical impulses) are sent down the axon of the motor neuron from the CNS and when the action potential reaches the end of the axon, hormones called neurotransmitters are then released. Neurotransmitters transport the electrical signal from the nerve to the muscle.
Muscle fiber types
The two main categories of muscle fiber are type I and type II—‘slow’ and ‘fast’ twitch, respectively. Type I fibers contain more blood cappilaries, higher levels of mitochondria (which transforms food into ATP) and myoglobin which allows for an improved delivery of oxygen. Since myoglobin is similar to hemoglobin (the red pigment which is found in red blood cells), type I fibers are also known as ‘red fibers.’ Type I fibers are also smaller in diameter and slower to produce maximal tension, but are also the most fatigue-resistant type of fiber.
Type II fibers have two subdivisions—IIa and IIx—based on their mechanical and chemical properties. Type II fibers are in many ways the opposite of type I fibers—they contain far fewer blood cappilaries, mitochondria and myoglobin. Since they have less myoglobin, they are not red, but white, which is why they are known as ‘white fibers.’ IIx fibers have a lower oxidative capacity and thusly tire out quicker. IIa, on the other hand, have a higher oxidative capacity and fatigue slower than IIx fibers (Herbison, Jaweed, and Ditunno, 1982; Tellis et al, 2012). IIa fibers are also known as intermediate fast twitch fibers since they can use both anarobic and aerobic metabolism equally to produce energy. So IIx fibers are a combo of I and II fibers. Type II fibers are bigger, quicker to produce maximal tension, and tire out quicker.
Now, when it comes to fiber typing between the races, blacks have a higher proportion of type II fibers compared to whites who have a higher proportion of type I fibers (Ama et al, 1986; Ceaser and Hunter, 2015; see Entine, 2000 and Epstein, 2014 for reviews). Higher proportions of type I fibers are associated with lower chance of cardiovascular events, whereas type II fibers are associated with a higher risk. Thus, “Skeletal muscle fibre composition may be a mediator of the protective effects of exercise against cardiovascular disease” (Andersen et al, 2015).
Now that the basics of muscle anatomy and physiology are apparent, hopefully the hows and whys of muscle contraction and what different muscle fibers do are becoming clear, because these different fibers are distributed between the races in uneven frequencies, which then leads to differences in sporting performance but also differents in health outcomes.
Muscle fibers and health outcomes
We now know the physiology and anatomy of muscle and muscle fiber typing. We also know the differences between each type of skeletal muscle fiber. Since the two races do indeed differ in the percentage of skeletal muscle fiber possessed on average, we then should find stark differences in health outcomes, part of the reason being these differences in muscle fiber typing.
While blacks on average have a higher proportion of type II muscle fibers, whites have a higher proportion of type I muscle fibers. Noting what I wrote above about the differences between the fiber types, and knowing what we know about racial differences in disease outcomes, we can draw some inferences on how differences in muscle fiber typing between races/individuals can then affect disease seriousness/acquisition.
In their review of black-white differences in muscle fiber typing, Ceaser and Hunter (2015) write that “The longitudinal data regarding the rise in obesity indicates obesity rates have been highest among non-Hispanic Black women and Hispanic women.” And so, knowing what we know about fiber type differences between races and how these fibers act when they fire, we can see how muscle fiber typing would contribute to differences in disease acquisition between groups.
Tanner et al (2001) studied 53 women (n=28, lean women; and n=25, obese women) who were undergoing an elective abdominal surgery (either a hysterectomy or gastric bypass). Their physiologic/anatomic measures were taken and they were divided into races: blacks and whites, along with their obesity status. Tanner et al found that the lean subjects had a higher proportion of type I fibers and a lower proportion of type IIx fibers whereas those who were obese were more likely to have a higher proportion of type IIb muscle fibers.
Like other analyses on this matter, Tanner et al (2001) showed that the black subjects had a higher proportion of type II fibers in comparison to whites who had a higher proportion of type I fibers (adiposity was not taken into account). Fifty-one percent of the fiber typing from whites was type I whereas for blacks it was 43.7 pervent. Blacks had a higher proportion of type IIx fibers than whites (16.3 percent for whites and 23.4 for blacks). Lean blacks and lean whites, though, had a similar percentage of type IIx fibers (13.8 percent for whites and 15 percent for blacks). It is interesting to note that there was no difference in type I fibers between lean whites and blacks (55.1 percent for whites and 54.1 percent for blacks), though muscle fibers from obese blacks contained far fewer type I fibers compared to their white counterparts (48.6 percent for whites and 34.5 for blacks). Obese blacks’ muscle fiber had a higher proportion of type IIx fibers than obese whites’ fiber typing (19.2 percent for whites and 31 percent for blacks). Lean blacks and lean whites had a higher proportion of type I fibers than obese blacks and obese whites. Obese whites and obese blacks had more type IIx fibers than lean whites and lean blacks.
So, since type II fibers are insulin resistant (Jensen et al, 2007), then they should be related to glucose intloerance—type II diabetes—and blacks with ancestry from West Africa should be most affected. Fung (2016, 2018) shows that obesity is a disease of insulin resistance, and so, we can bring that same rationale to racial differences in obesity. Indeed, Nielsen and Christensen (2011) hypothesize that the higher prevalence of glucose intolerance in blacks is related to their lower percentage of type I fibers and their higher percentage of type II fibers.
Nielsen and Christensen (2011) hypothesize that since blacks have a lower percentage of type I fibers (the oxidative type), this explains the lower fat oxidation along with lower resting metabolic rate, sleeping metabolic rate, resting energy expenditure and Vo2 max in comparison to whites. Since type I fibers are more oxidative over the glycolitic type II fibers, the lower oxidative capacity in these fibers “may cause a higher fat storage at lower levels of energy intake than in individuals with a higher oxidative capacity” (Nielsen and Christensen, 2011: 611). Though the ratio of IIx and IIa fibers are extremely plastic and affected by lifestyle, Nielsen and Christensen do note that individuals with different fiber typings had similar oxidative capacity if they engaged in physical activity. Recall back to Caesar and Hunter (2015) who note that blacks have a lower maximal aerobic capacity and higher proportion of type II fibers. They note that lack of physical activity exacerbates the negative effects that a majority type II fibers has over majority type I. And so, some of these differences can be ameliorated between these two racial groups.
The point is, individuals/groups with a higher percentage of type II fibers who do not engage in physical activity have an even higher risk of lower oxidative capacity. Furthermore, a higher proportion of type II fibers implies a higher percentage of IIx fibers, “which are the least oxidative fibres and are positively associated with T2D and obesity” (Nielsen and Christensen, 2011: 612). They also note that this may explain the rural-urban difference in diabetes prevalance, with urban populations having a higher proportion of type II diabetics. They also note that this may explain the difference in type II diabetes in US blacks and West African natives—but the reverse is true for West Africans in the US. There is a higher rate of modernization and, with that, a higher chance to be less physically active and if the individual in question is less physically active and has a higher proportion of type II fibers then they will have a higher chance of acquiring metabolic diseases (obesity is also a metabolic disease). Since whites have a higher proportion of type I fibers, they can increase their fat intake—and with it, their fat oxidation—but this does not hold for blacks who “may not adjust well to changes in fat intake” (Nielsen and Christensen, 2011: 612).
Nielsen and Christensen end their paper writing:
Thus, Blacks of West African ancestry might be genetically predisposed to T2D because of an inherited lower amount of skeletal muscle fibre type I, whereby the oxidative capacity and fat oxidation is reduced, causing increased muscular tissue fat accumulation. This might induce skeletal muscle insulin resistance followed by an induced stress on the insulin-producing beta cells. Together with higher beta-cell dysfunction in the West African Diaspora compared to Whites, this will eventually lead to T2D (an overview of the ‘skeletal muscle distribution hypothesis’ can be seen in Figure 2).
Lambernd et al (2012) show that muscle contractions eliminated insuin resistance by blocking pro-inflammatory signalling pathways: this is the mechanism by which physical activity decreases glucose intolerance and thusly improves health outcomes—especially for those with a higher proportion of type II fibers. Thus, it is important for individuals with type II fibers to exercise, since sedentariness is associated with an age-related insulin resistance due to impaired GLUT4 utilization (Bunprajun et al, 2013).
(Also seeMorrison and Cooper’s (2006) hypothesis that “reduced oxygen-carrying capacity induced a shift to more explosive muscle properties” (Epstein, 2014: 179). Epstein notes that the only science there is on this hypothesis is one mouse and rat study showing that low hemoglobin can “induce a switch to more explosive muscle fibers” (Epstein, 2014: 178), but this has not been tested on humans to see if it would hold. If this is tested on humans and if it does hold, then that would lend credence to Morrison’s and Cooper’s (2006) hypothesis.)
Conclusion
Knowing what we know about muscle anatomy and physiology and how muscles act we can understand the influence the different muscle types have on disease and how they contribute to disease variation between race, sex and the individual level. Especially knowing how type II fibers act when the individual in question is insulin resistant is extremely important—though it has been noted that individuals who participate in aerobic exercise decrease their risk for cardiometabolic diseases and can change the fiber distribution difference between IIx and IIa fibers, lowering their risk for acquiring cardiometabolic diseases (Ceaser and Hunter, 2015).
Thinking back to sarcomeres (the smallest contractile unit in the muscle) and how they would act in type II fibers: they would obviously contract much faster in type II muscles over type I muscles; they would then obviously tear faster than type I muscles; since type II muscles are more likely to be insulin resistant, then those with a higher proportion of type II fibers need to focus more on aerobic activity to “balance out” type IIx and IIa fibers and decrease the risk of cardiometabolic disease due to more muscle contractions (Lambernd et al, 2012). Since blacks have a higher proportion of type II fibers and are more likely to be sedentary than whites, and since those who have a higher proportion of type II fibers are more likely to be obese, then it is clear that exercise can and will ameliorate some of the disparity in cardiometabolic diseases between blacks and whites.
Racial differences in body fat are clear to the naked eye: black women are more likely to carry more body fat than white women; Mexican American women are more likely to carry more body fat than white women, too. Different races/ethnies/genders of these races/ethnies have different formulas to assess body fat through the use of skin-folds. The sites to grasp the skin is different based on gender and race.
Body mass index (BMI) and waist circumference is overestimated in blacks, which means that they need different formulas to assess their BMI and adiposity/lean mass. Race-specific formulas/methods are needed to assess body fat and, along with it, disease risk, since blacks are more likely to be obese (black women, at least, it’s different with black American men with more African ancestry, see below). The fact of the matter is, when matched on a slew of variables, blacks had lower total and abdominal fat mass than whites.
This is even noted in Asian, black and white prepubertal children. He et al (2002) show that sex differences in body fat distribution are present in children who have yet to reach puberty and the differences in body fat in Asians is different than that from blacks and whites which also varies by sex. Asian girls had greater gynoid fat by DXA scan only, with girls having greater gynoid fat than boys. Asian girls had lower adjusted extremity fat and gynoid fat compared to white and black girls. Though, Asian boys had a lower adjusted extremity by fat as shown by DXA (a gold standard in body fat measurement) when compared to whites, but greater gynoid fat than whites and blacks.
Vickery, Cureton, and Collins (1988) argue that, if accurate estimates of body fat percentages are to be obtained, race-specific formulas need to be developed and used as independent variables to assess racial differences in body fat percentage. Differences in muscularity don’t seem to account for these skinfold differences, nor does greater mesomorphy. One possible explanation for differences in skinfold thickness is that blacks may store most of their body fat subcutaneously. (See Wagner and Heyward, 2000 for a review on fat patterning and body composition in blacks and whites.)
The often-used Durnin-Womersley formula which is used to predict body fat just from skin folds. However, “The 1974 DW equations did not predict %BF(DXA) uniformly in all races or ethnicities” (Davidson et al, 2011). Truesdale et al (2016) even show that numerous formulas used to estimate percent body fat are flawed, even some formulas used on different races. Most of the equations tested showed starkly different conclusions. But, this is based on NHANES data and the only data they provide regarding skin-folds is the tricep and subscapular skinfold so there may still be more problems with all of the equations used to assess body fat percentage between races. (Also see Cooper, 2010.)
Klimentidis et al (2016) show that black men—but not black women—seem to be protected against obesity and central adiposity (fat gain around the midsection) and that race negatively correlated with adiposity. The combo of male gender and West African ancestry predicted low levels of adiposity compared to black Americans with less African ancestry. Furthermore, since black men and women have—theoretically—the same SES, then cultural/social factors would not play as large a role as genetic factors in explaining the differences in adiposity between black men and black women. Black men with more African ancestry had a lower WHR and less central adiposity than black men with less African ancestry. If we assume that they had similar levels of SES and lived in similar neighborhoods, there is only one reason why this would be the case.
One interpretation is that AAs are exposed to environmental and/or cultural factors that predispose them to greater obesity than EAs. Possibly, some of the genes that are inherited as part of their West-African ancestry are protective against obesity, thereby “canceling out” the obesifying effects of environment/culture, but only in men. Another interpretation is that genetic protection is afforded to all individuals of African descent, but this protection is overwhelmed by cultural and/or other factors in women.
Black men do, as is popularly believed, prefer bigger women over smaller women. For example, Freedman et al (2004) showed that black American men were more likely to prefer bigger women. Black American men “are more willing to idealize a woman of a heavier body size, with more curves, than do their White American counterparts” (Freedman et al, 2004: 197). It is then hypothesized that black American men find these figures attractive (figures with “more curves” (Freedman et al, 2004: 197))to protect against eating pathologies, such as anorexia and bulimia. So, it has been established that black men have thinner skin folds than whites which leads to skewed lean mass/body fat readings and black men with more African ancestry are less likely to be obese. These average differences between races, of course, contribute to differing disease acquisition.
I have covered differences in body fat in a few Asian ethnies and have come to the obvious conclusion: Asians, at the same height, weight etc as whites and blacks, will have more adipose tissue on their bodies. They, too, like blacks and whites, have different areas that need to be assessed for skin folds to estimate body fat.
Henriques (2016: 29) has a table on the equations for calculating estimated body density from skin fold measures from various populations. Of interest are the ones on blacks or ‘Hispanics‘, blacks or athletes and blacks and whites. (The table is provided from NSCA, 2008 so the references are not in the back of the text.)
For black and ‘Hispanic’ women aged 18-55 years, the sites to use for skin-folds are the chest, abdomen, triceps, subscapular, suprailiac, midaxillary, and the thigh. For blacks or athletes aged 18-61 years, the sites to use are the same as before (but a different equation is used for body fat estimation). For white women or anorexic women aged 18-55, the sites used are just triceps, suprailiac and the thigh. For black and white boys aged 6-17, only the triceps and the calf is used. It is the same for black and white girls, but, again, a different formula is used to assess body fat (Henriques, 2016: 29).
Morrison et al (2012) showed that white girls had a higher percent body fat when compared to black girls at ages 9-12 but every age after, black girls had higher percent body fat (which is related to earlier menarche in black girls since they have higher levels of body fat which means earlier puberty; Kaplowitz, 2008). Black girls, though, had higher levels of fat in their subscapular skin folds than white girls at all ages.
So, it seems, there are population-/race-specific formulas that need to be created to better assess body fat percentage in different races/ethnies and not assume that one formula/way of assessing body fat should be used for all racial/ethnic groups. According to the literature (some reviewed here and in Wagner and Heyward, 2000), these types of formulas are sorely needed to better assess health markers in certain populations. These differences in body fat percentage and distribution then have real health consequences for the races/ethnies in question.